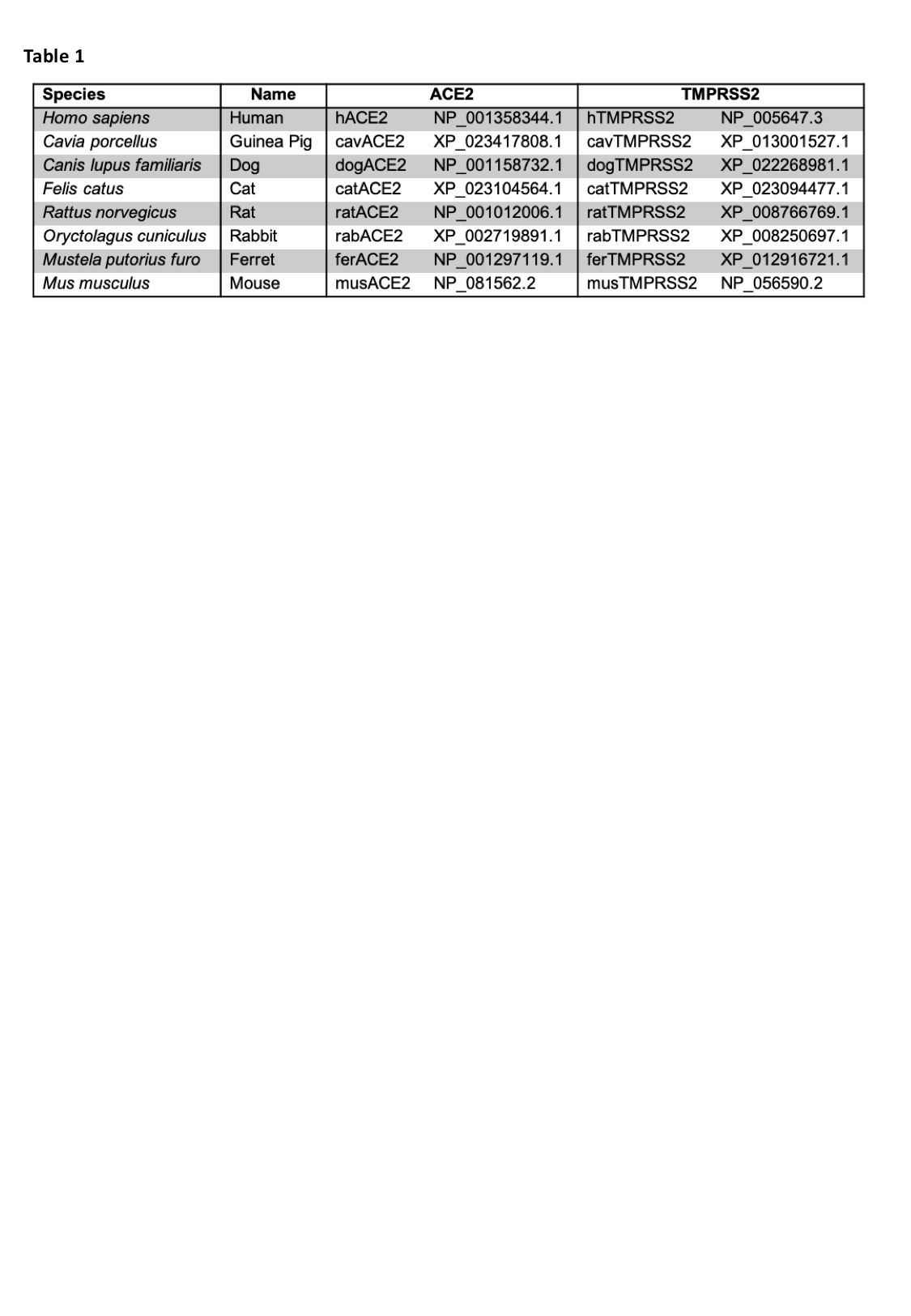
ACE2 interaction with the SARS-Cov-2 spike protein differs between species
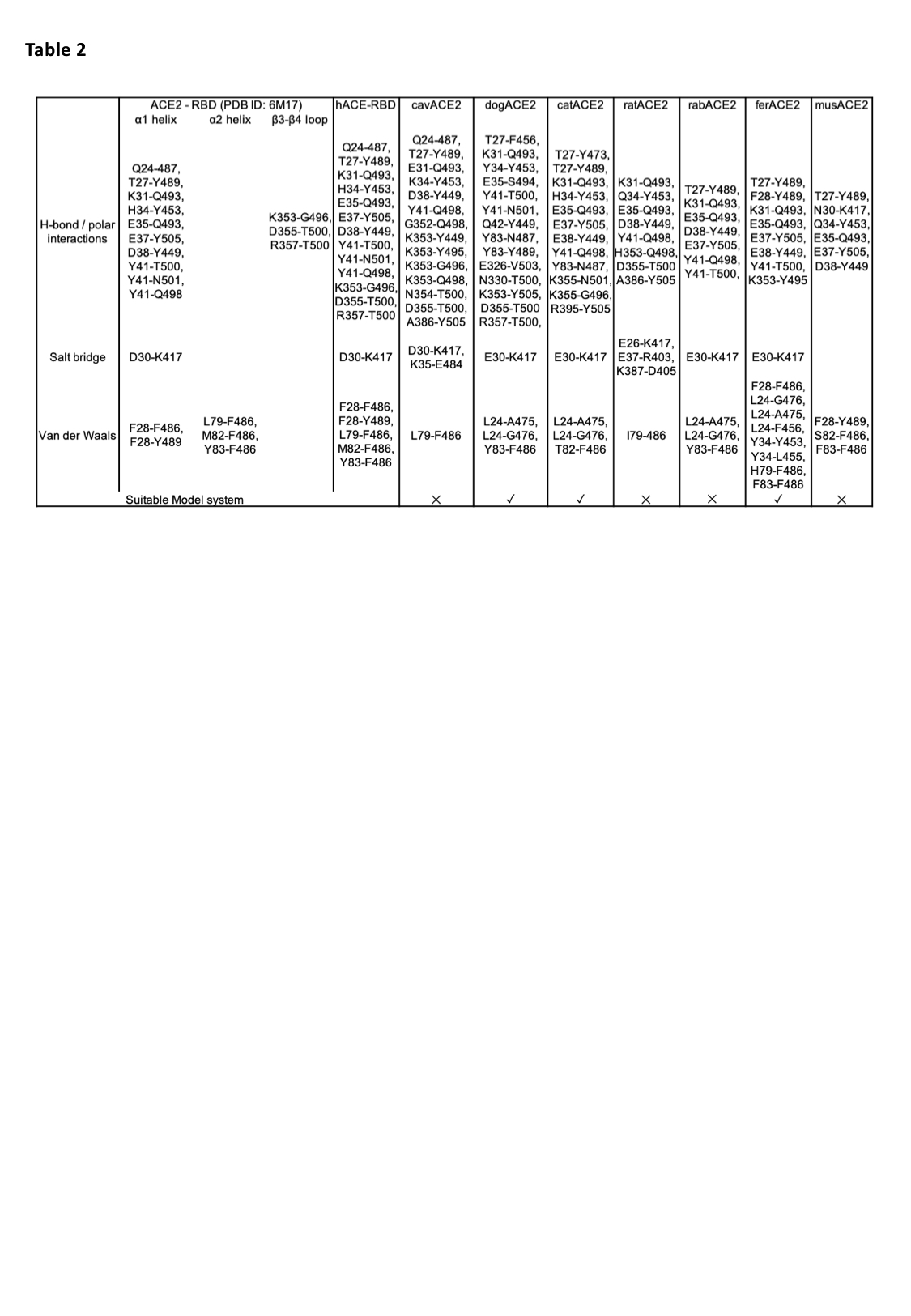
ACE2 is a zinc carboxypeptidase type I transmembrane protein, with an extracellular N-terminal peptidase domain (PD) and a cytosolic C-terminal collectrin-like domain (CLD) (Figure S1). The receptor binding domain (RBD) of the SARS-CoV-2 S protein binds directly to the ACE2 PD, and analysis of the crystal structure of this complex shows that the interaction is mostly driven by polar interactions (Table 2). Of particular interest are the hydrogen bond between ACE2 E35 and S protein RBD Q493, the salt bridge between ACE2 D30 and RBD K417, and the hydrophobic interaction between ACE2 M82 and RBD F486. Interestingly, Q493, K417 and F486 are not conserved between SARS-CoV and SARS-CoV-2, and these differences are linked to the higher affinity of the SARS-CoV-2 S protein for ACE2 [30].
In order to predict if the SARS-CoV-2 S protein binds ACE2 of other animal species, focusing mostly on laboratory model systems, we generated homology models for cavACE2, dogACE2, catACE2, ratACE2, rabACE2, ferACE2, musACE2 (Table1, Figure 1A). Alignment of the ACE2 sequences from these species, revealed a high conservation, with a sequence identity between 77.5 % and 85.2% (Supplemental Figure S1-2). This allowed us to produce reliable ACE2 PD models via homology modelling using the hACE2 as a template. We then ran docking simulations between the ACE2 PD models and the SARS-CoV-2 S protein RBD to generate optimised complexes. A docking simulation using hACE2 as a control was also performed. The presence of a similar network of interactions in the docking output for the hACE2 simulation compared to the one observed in the EM structure was used to validate the approach adopted (Table 2).
Overall the hydrophobic contributions that stabilise the RBD-ACE2 complex are similar in all models, with the ferACE2-RBD having a slightly higher number of hydrophobic contacts (Table 2). Interestingly, M82 in the hACE2 is not conserved across species (Supplemental Figure S1), and only the catACE2 and musACE2 form van der Waals interaction between residue 82 and the RBD F486 (Supplemental Figure S3). However, RBD F486 is in contact, in all complex structures, with a relatively hydrophobic patch formed by the ACE2 residues 28, 79 and 83 (Table 2 and Supplemental Figure S3). Comparison of the surface electrostatic potentials of the ACE2 models identified a similar distribution of charges on the α1 helix, α2 helix and β3-β4 loop across species (Figure 1A).
Differences between the structure of hACE2 and musACE2 have been previously described to explain why SARS-CoV is a mild infection in mice [31]. The most strikingly difference between the hACE2 and musACE2 are the D30 to N30 and K31 to N31 substitutions. This results in the lack of salt bridges and concomitant lower number of H-bonds in the musACE2-SARS-CoV-2 RBD complex (Table 2). Specifically, the salt bridge with the K417 of the RBD seems to be a major driver of the interaction. In fact, similar to the musACE2, the ratACE2 has an Asn in position 30, which prevents formation of a salt bridge with K417 in the RBD. However, E26 in the ratACE2 forms a salt bridge with K417, resulting in a slightly altered complex structure with a shift of 6.5 Å of the RBD over the ratACE2, compared to its relative position in the human complex (Figure 1B). Furthermore, substitution of M82 to N82 in the ratACE2 introduces an N-glycosylation site [32], which may create steric clashes with F486 and N487. Taken together, the differences in binding mode would suggest that mice and rat are unsuitable models for the study of COVID-19. Similarly, the presence of a salt bridge between K35 and E484 in the cavACE2-RBD complex would make guinea pig an unsuitable model for the study of inhibitory antibodies and small molecules targeting the ACE2
- SARS-CoV-2 S protein interaction.
SARS-CoV S protein in complex with ACE2
In order to further validate the approach adopted we carried out docking simulations between SARS-CoV RBD, for which more experimental data are available, and hACE2, cavACE2, dogACE2, catACE2, ratACE2, rabACE2, ferACE2, musACE2. Indeed, comparison of the SARS-CoV and the SARS-CoV-2 RBD in complex with ACE2 shows that the two co- crystal structures are comparable (RMSD 2AJF) and the binding interfaces are similar (Table 3). In line with previously published data, we see that the SARS-CoV S has a smaller interaction surface and a lower number of interactions with ACE2 compared to the SARS- CoV-2 S protein (Table 3) [33].
The mode of binding of SARS-CoV RBD to hACE2 has several differences compared to that of the other ACE2 proteins analysed. While the binding of RBD to hACE2 is driven by polar interactions, similarly to what we observed for SARS-CoV-2, in all other SARS-CoV RBD-ACE2 models there are fewer H-bonds and a concomitant increase in hydrophobic interactions. Importantly, the substitution H34 to Y/L34 introduces a steric interference, which results in a shift of ~3 Å of the 441-456 loop of RBD bound to the dogACE2 and the cavACE2, compared to its relative position in the human complex. Similarly, the substitution of E329 with T/A/Q/K329 prevents the formation of a salt bridge with R426, and in the cavACE2 complex creates a charge repulsion. Overall, this would suggest a lower affinity of the RBD for the cavACE2, dogACE2, catACE2, ratACE2, rabACE2, ferACE2, musACE2, in line with previously published data showing different susceptibility to infection of animal models [34].
Indeed, SARS infection in cats, ferrets, mice, guinea pigs, and rats is weaker and does not replicate the human disease in all its aspects [34]. Taken together our approach may provide a rational for the observed experimental differences of the infection in human and animal models.
TMPRSS2 is highly conserved across species
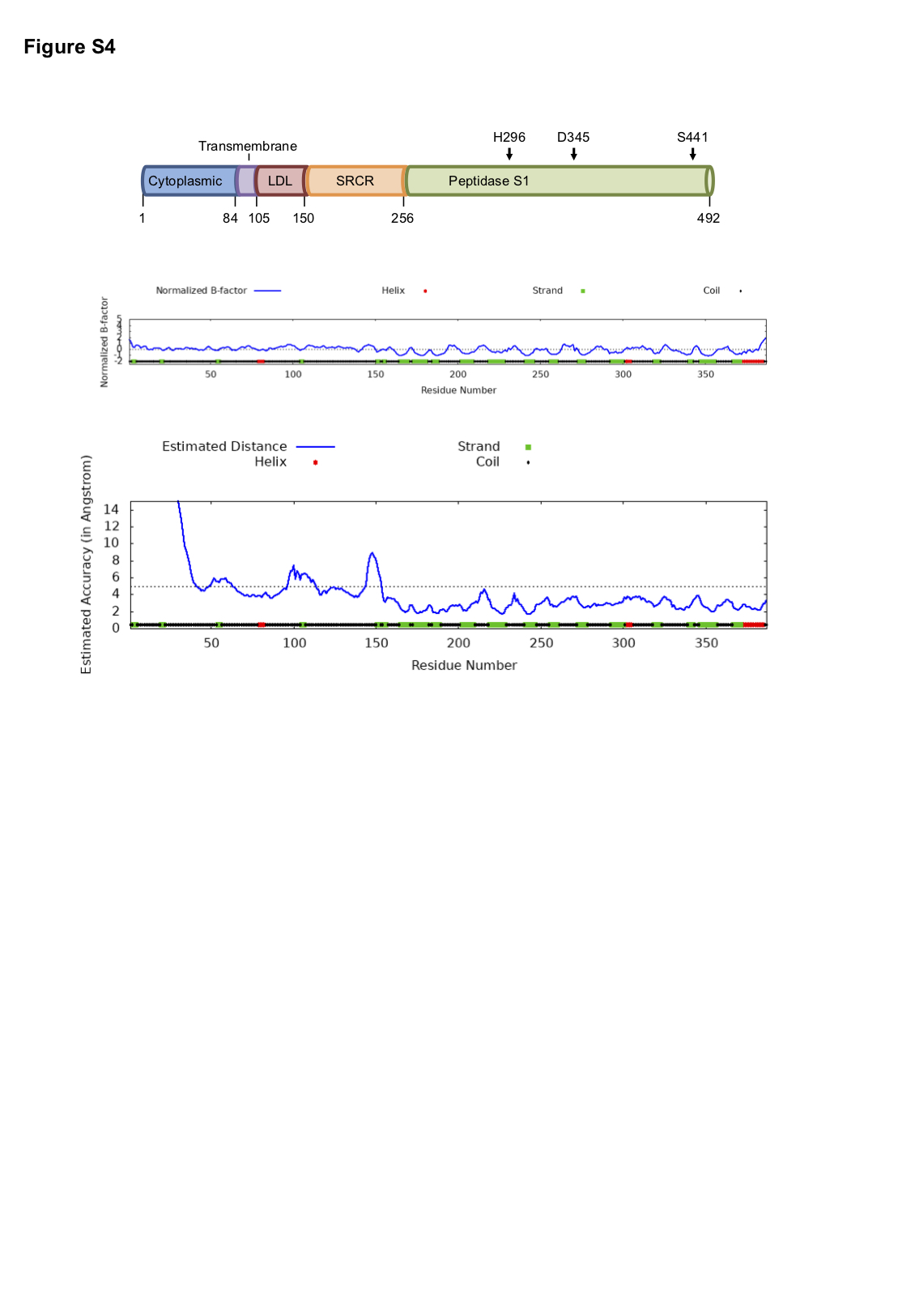
TMPRSS2 is a type II transmembrane serine protease (TTSPs), with an extracellular region composed of a low-density lipoprotein (LDL) receptor class A domain, a scavenger receptor cysteine-rich (SRCR) domain and a peptidase S1 domain containing the catalytic triad (Supplemental Figure S4). TMPRSS2, similar to other TTSPs, has high affinity towards substrates containing an Arg residue in the P1 position. Indeed, TMPRSS2 recognises the SPRRAR/SVASQS sequence in the SARS-CoV-2 S glycoprotein and cleaves S1 from S2 between residues 685/686 [35].
In order to predict if TMPRSS2 from other animal species can cleave the SARS-CoV-
2 S protein, the extracellular domain sequences of hTMPRSS2, cavTMPRSS2, dogTMPRSS2, catTMPRSS2, ratTMPRSS2, rabTMPRSS2, ferTMPRSS2 and musTMPRSS2 were aligned. The alignment revealed high conservation with a sequence identity between 75.11% and 83.97 % (Supplemental Figure S2). We then generated a model for hTMPRSS2 (Figure 2A and S4) using I-TASSER, and the best model had a C-score of -
0.52 with a TM-score of 0.65 ± 0.13 and an RMSD of 7.9 ± 4.4 Å. The model has the three conserved disulphide bonds on the peptidase S1 domain, characteristic feature of all TTSPs, between residues C281-C297, C410-C426 and C437-C465 [36]. Disulphide bonds are also present between C113-C126, C120-C139, C133-C148, C172-C231 and C185-C241, which further validates the reliability of the models generated (Figure 2A and S4).
The pocket containing the catalytic triad has a uniform negative charge, which favours electrostatic interactions with the Arg reach peptide of the S protein (Figure 2B). Using this structural information, we identified the residues surrounding the catalytic triad that form a pocket on the head of TMPRSS2. Interestingly, this pocket is identical in all species studied and no substantial differences from the hTMPRSS2 were observed (Figure 2C). Taken together these data suggest that cavTMPRSS2, dogTMPRSS2, catTMPRSS2, ratTMPRSS2, rabTMPRSS2, ferTMPRSS2, musTMPRSS2 can cleave the SARS-CoV-2 S glycoprotein in a similar way to hTMPRSS2.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}