Study design
This is a multicentre, randomised, controlled trial to evaluate the efficacy and safety of JTW in patients with depression. A flowchart for the study is shown in Fig 1. The Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) checklist is shown in Supplemental file 1.
Qualified patients will be randomised into either the treatment or the control group (in a 1:1 ratio).Recruited patients will receive JTW plus fluoxetine or fluoxetine alone once a day for eight weeks. After the 8-week treatment period, the patients will be followed up for four weeks. The outcome measures include the Hamilton Depression(HAMD) Rating Scale scores, TCM syndrome integral scale scores, Wisconsin Card Sorting Test (WCST), blood metabonomics, urine metabonomics and brain structure and function on fMRI.
Participants
Inclusion criteria
To participate in the study, patients should meet the following criteria:
(1)Conform to depression diagnostic criteria of the Diagnostic and Statistical Manual of Mental Disorders’ American psychiatric diagnosis standard’,4thEdition (DSM-IV);
(2)Conform to the diagnostic criteria of TCM syndrome of disharmony between the heart and kidney;
(3)HAMD 24-item score of ≥20 points and <35 points;
(4)Age, 18–65 years;
(5)Total HAMA score of ≤21 points, depressed mood (item 6) score ≥2 points and anxious mood (item 1) score <3 points;
(6)Not taking antidepressants or any other medicine but stopping for two weeks;
(7)Sign the informed consent form.
Exclusion criteria
Patients with the following criteria will be excluded:
(1)Secondary to other mental or physical illness and depression with severe psychotic symptoms;
(2)Severe anxiety (HAMA total score >21 points);
(3)Allergic constitution or previous research on drug allergy;
(4)Suicidal thoughts;
(5)Bipolar disorder, refractory depression;
(6)Intracranial cerebrovascular disease, neurodegenerative diseases, intracranial tumour, high blood pressure, or diabetes, leading to brain vascular distribution or abnormal blood flow of systemic disorders;
(7)Serious diseases of other systems or severe heart, liver and renal insufficiency;
(8)Glaucoma and epilepsy;
(9)Pregnancy or lactation or quasi-pregnancy;
(10)Positive urine pregnancy test in women of childbearing age;
(11)Contraindications on MR inspection and those who cannot complete MR scan or cannot adapt to the environment and noise of the MRI machine;
(12)Participation in or planning to participate in other observational drug studies within 30 days;
(13)Poor compliance or inability to interview regularly.
Ethics and recruitment
All patients will sign informed consent forms prior to inclusion. The study has been approved by the Ethics Committee of Tianjin University of Traditional Chinese Medicine. Any revisions in the study protocol will be submitted to the Ethics Committee.
Through advertisements and referrals, a total of 40 qualifying patients will be recruited from two research centres: the First Teaching Hospital of Tianjin University of Traditional Chinese Medicine and the Tianjin Academy of Traditional Chinese Medicine Affiliated Hospital.
Sample size
In reference to the relevant literature, the specific process to estimate the sample content of each group is as follows[12–14]. According to the sample size estimation formula for clinical trials,
[Due to technical limitations, this equation is only available as a download in the supplemental files section.]
where n is the number of cases required for each group, μ1 and μ2 are the expected mean of the treatment and control groups, respectively, σ2 is the standard deviation of the control group, and ƒ (α, β) is taken as 10.5. The HAMD score is considered the primary outcome. From previous studies [15–17], considering the mean, we get μ1 = 9.39 and μ2 = 12.49.The standard deviation of the control group is 2.97, and putting the above data into the formula, n was calculated to be19.2.Using the calculation, the sample size in each group was determined to be about20; therefore, the total sample size was 40, with 20 in each group.
Randomisation
Treatment allocation will be done when participants meet the inclusion criteria and sign the informed consent form. Patients will be randomly divided into two groups, the treatment and control groups, with a 1:1 distribution ratio according to the random numbers generated by IBM SPSS Statistics software version 19.0.The recruiter will obtain a sequence number from the distributor when a patient is eligible for the trial. To prevent subjective investigator bias from affecting the authenticity of the clinical research results, the confidential randomisation method uses a closed envelope method.
Interventions
Patients randomised to the treatment group will be administered one bag of JTW granules(C.chinensis Franch 15g, Ci.cassia Presl 2g) dissolved in warm water once daily plus fluoxetine 20mg once daily for eight weeks. Patients in the control group will receive fluoxetine 20mg once daily for eight weeks.JTW granules are manufactured by Beijing Tcmages Pharmaceutical Co., Ltd, and fluoxetine is manufactured by Eli Lilly and Company.
Outcome assessment
Primary outcome
HAMD scores: This study uses 24-item HAMD scale, which is summarised as seven categories of factors and can be more concise and clearly reflect the patient characteristics. The seven types of depression scale factors include the following: 1) anxiety/somatisation factor, 18 points; 2) weight: 2 points; 3) cognitive disturbance factor, 21 points; 4) day and night change factor, 2 points; 5) retardation, 14 points; 6) sleep dysfunction factor, 6 points and 7) withdrawal and self-accusation, 12 points. Factor scores can reflect the characteristics of patients with depression symptoms as well as the characteristics of the changes before and after the drug interventions.
Secondary outcome
- TCM syndrome integral scale scores: TCM symptoms of all patients will be assessed through the TCM syndrome integral scale.
- WCST: Patients were assessed for cognitive function using the computer version of WCST.WCST consists of four templates and 48 cards, which are used to assess the cognitive function of patients by the total number of WCST, number of correct responses, total number of errors, number of persistent errors, and number of random errors.
- Blood and urine metabonomics: To systematically analyse the metabolic status of patients with depression and study the metabolomics characteristics of depression and metabolic changes after treatment.
- Brain structure and function on fMRI: MRI can reveal brain tissue abnormalities in patients with depression in the aspects of brain microstructure, brain function changes and changes in brain metabolites, and fMRI combines the three aspects of function, anatomy and imaging[18–20]. Diffusion tensor imaging (DTI) technique is used to detect differences and changes in fractional anisotropy values in the whole brain. The changes in cerebral white matter integrity in patients with depression before and after treatment will be compared to study the effect of JTW on brain metabolites and the changes in brain metabolites by magnetic resonance spectroscopy.
Safety reporting
Adverse events (AEs) are defined as any unfavourable and unintended sign, symptom or disease arising in participants in either group during the trial period, regardless of their relation to the experimental treatment. The investigator should take appropriate measures to ensure participants’ safety and follow-up the outcome of any AEs. Researchers must fill out relevant information for AEs in the safety evaluation form. The record includes a detailed description of AEs, time of occurrence, time of suspension, duration (which can be recorded in days or hours), severity and frequency of the occurrence, manner of treatment, amount of treatment and course of treatment as well as the causal analysis of AEs and trial treatment. All clinical data for AEs should be recorded on case report forms (CRFs).
Data collection, management and quality control
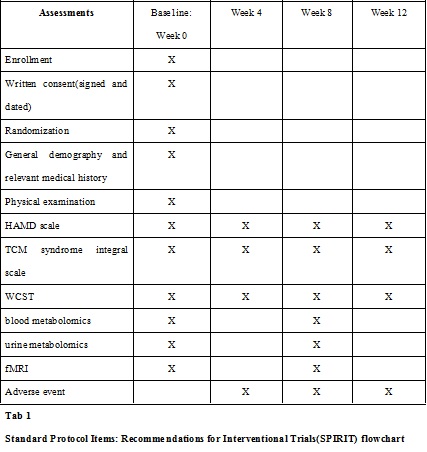
A CRF will be used for each participant to collect relevant data, and participants will be evaluated every four weeks during the study. Data on HAMD, WCST and TCM syndrome integral scale will be collected at each visit (week 0 ± 3 days, week 4 ± 3 days, week 8 ± 3 days and week 12 ± 3 days). Blood metabolomics, urine metabolomics and fMRI will be measured only at the first and third visits (week 0 ± 3 days and week 8 ± 3 days, respectively). The SPIRIT flowchart of the trial is shown in Table1.
After review by clinical inspectors, completed CRFs will be sent to a specified statistics centre, and data entry and management will be completed by two individual data administrators to ensure data accuracy. All documents collected in this study will be stored securely. Participants will be referred to by a specific randomisation number rather than by their names in all other documents in this study, except the informed consent form.
The trial group developed a quality inspection system, and the trial set up quality inspector. The details of the quality inspector’ implementation include examining the experimental protocols and procedures by the test operators and recording and reporting all research data and the truthful, accurate and complete CRFs, while assuring that the original data are consistent.
Data analysis
The primary analysis is described below. Balance comparison: To compare the demographics and other basic indicators to measure the comparability of the two groups. Analysis of validity: To calculate the HAMD scores of the patients in the two groups during the treatment period and to determine the mean and standard deviation of the values at baseline from those after treatment. Analysis of action mechanism: To calculate the TCM syndrome integral scale scores, WCST, Blood and urine metabonomics, and to analyse MRI image of the patients in the two groups. Statistical analysis or Pearson correlation coefficient was performed using analysis of variance (ANOVA).
Statistical analysis will be conducted as an intention-to-treat analysis. Statistical analysis will be performed using SPSS Statistics version17.0 software. The statistical significance is defined as a two-sided P-value of <0.05. Baseline differences among the groups will be assessed using 1-factor ANOVA for measurement data and the χ2 test for enumeration data. In addition, χ2 test will be used for categorical variables. The changes in scores from baseline to endpoint of treatment will be assessed using a paired t test for measurement data and a signed rank test for enumeration data. Comparisons between groups will be conducted using ANOVA and a rank test.
The researchers conducting the data analysis will not be involved in experimental or clinical decision-making processes to prevent any bias caused by subjective factors from the researchers.
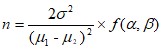{kind=link}
{kind=link}