Dupuytren's disease is a common localized fibrotic disorder of the palmar fascia that affects 3% to 5% of the UK population (Gerber et al. 2011). The pathogenesis of Dupuytren's disease remain largely unknown (Karbowiak et al. 2021). Recent research using single-cell RNA sequencing (scRNA-seq) has expanded our understanding of the main cellular and molecular processes in mesenchymal cells that drive Dupuytren's disease (Dobie et al. 2022). However, the involvement of other cells in developing Dupuytren's disease is largely unresolved. Endothelial cells (ECs) play an important role in the pathophysiology of some fibrotic diseases, such as lung fibrosis, keloid, and systemic sclerosis, according to growing studies (Xu et al. 2016; Liu et al. 2022; Apostolidis et al. 2018). In this study, scRNA-seq analysis from Dupuytren's disease, healthy dermis (DE), and nonpathogenic (Skoog's) fascia (SF) were performed to explore the major pathogenic ECs subpopulations associated with Dupuytren's disease.
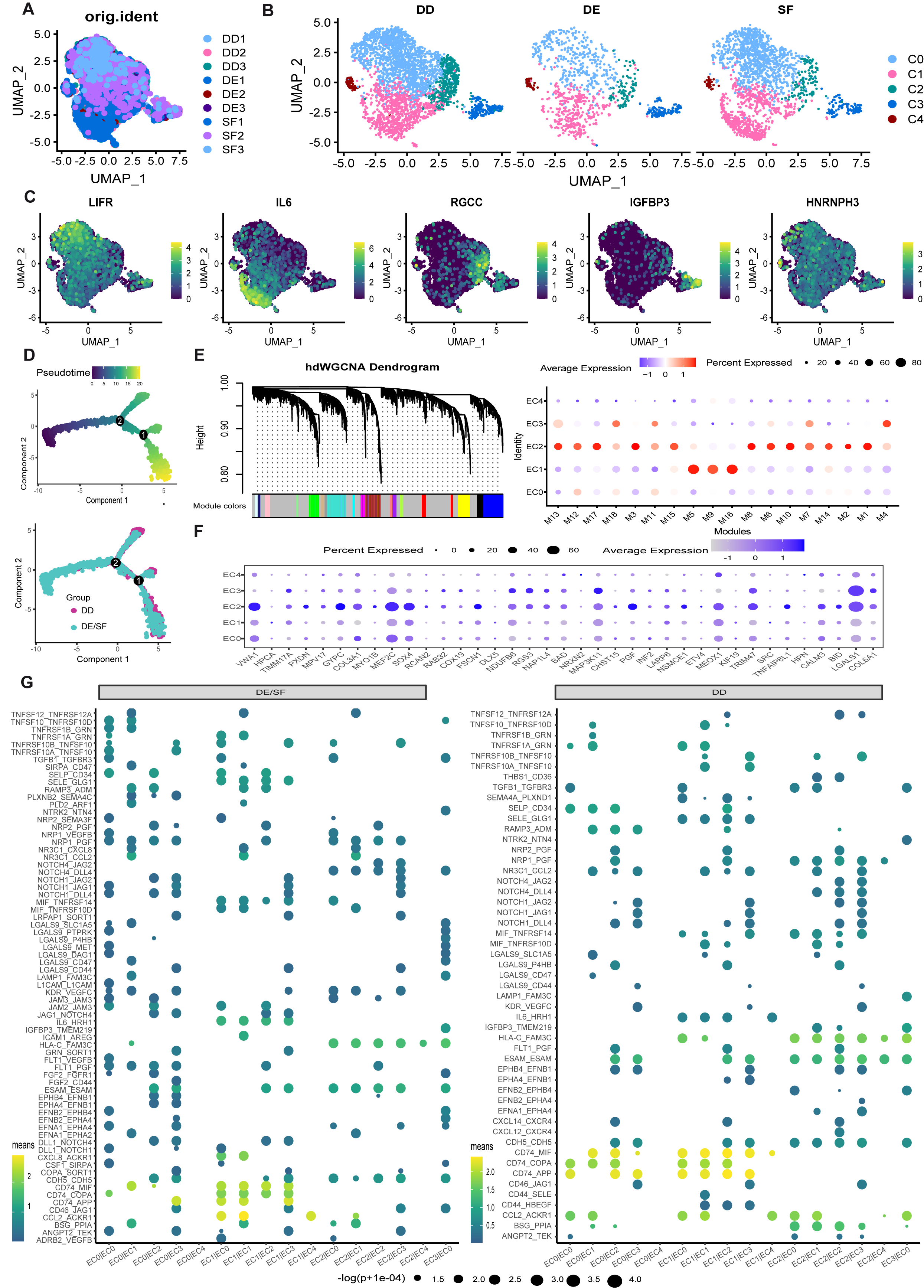
In order to reveal the cellular composition and functional status of ECs, different ECs populations were identified using an unsupervised clustering approach embedded in Seurat (Figs. 1A and Supplementary Fig. 1A-B), as described on the Supplementary Methods. Five unique clusters, EC0 (LIFR + ECs), EC1 (IL6 + ECs), EC2 (RGCC + ECs), EC3 (IGFBP3 + ECs), and EC4 (HNRNPH3 ECs) were found based on the gene expression profile (Figs. 1B and Supplementary Fig. 1C). Contrary to DE and SF, patients with Dupuytren's disease accounted for a greater percentage of EC2 (p < 0.05, permutation test) (Fig. 1C-D). However, no statistically significant differences in other ECs subpopulations were detected between Dupuytren's disease and healthy controls (DE/SF). The use of RNA velocity analysis to derive transcriptional dynamics and estimate the future states of a cell is a valuable tool for investigating cell fate trajectories. Our analysis revealed the possibility of differentiation from EC1 to EC2 (Fig. 1E). To further validate transcriptional properties at different stages of ECs development, Monocle2 was used to perform a pseudotime analysis to identify gene expression trajectories connected with functional changes. In accordance with the findings of the RNA velocity analysis, EC1 was found to be mostly at the start of the developmental trajectory, whereas EC2 was found to be primarily at the conclusion of the pseudo-temporal trajectory (Fig. 1F and Supplementary Fig. 1D). Notably, we observed higher expression of genes involved in positive regulation of cell population proliferation, such as AREG and VEGFC, at the beginning of the developmental trajectory, while genes involved in immune response regulation, such as BTNL9 and CD34, were prominent at the end of the trajectory (Fig. 1G-H). The differently expressed genes between Dupuytren's disease and healthy controls (DE/SF) and gene ontology (GO) analysis were undertaken to investigate the impact of disease-related alterations. Our findings revealed significant upregulation of genes involved in the inflammatory response to wounding, such as TIMP1 and TGFB1, as well as genes involved in the positive regulation of the canonical Wnt signaling pathway in EC2, suggesting that patients with Dupuytren's disease had a more inflammatory activation state than healthy controls (Fig. 1I-J). In order to identify the main molecular characteristics of EC2, a high-dimensional weighted correlation network analysis (hdWGCNA) was performed. After generating metacells of the ECs dataset, several modules that are specific for EC2 and diseases were found (Fig. 1K-L and Supplementary Fig. 1E). For example, hub genes of the disease-downregulated M2 including TIMP1, PRCP, and RHOA, are consistent with its enrichments of GO terms related to wound healing. Interestingly, CD99 was found to be an important hub gene in M2 and to have increased levels in Dupuytren's disease (Fig. 1L). Further experiments are warranted to investigate its role in the diseases.
Typically, the transcriptional profile is substantially determined by the regulation of several major transcription factors (TFs). The single-cell regulatory network inference and clustering for pySCENIC analysis was then used to predict TFs contribute to the establishment and maintenance of transcriptional characterizations across different ECs subpopulations. Based on activity scores, a regulon specificity score (RSS) was assigned to each regulon in each subpopulations, and the regulons with the highest RSS values were chosen as major regulators. Our analysis revealed that the most specific regulons in endothelial cells are CREB3L1(+), ETV4(+), FOXD1(+), and GTF3A(+), which is substantiated by considerably higher activity scores across cells in EC2 (Figs. 1M). Notably, ETV4 has been recognized as a critical regulator of the angiogenic response in ECs (Harel et al. 2021). pySCENIC also predicted several downstream target genes, for example, SOX4 is a significant downstream target within the regulon of the EC2-specific TF ETV4 (Figs. 1N). Taken together, the reports from literature mining significantly support the reliability of our gene regulatory network (GRN) inference analysis and establish the foundation for future research into the role of TFs in ECs pathology.
To thoroughly investigate interactions in ECs, we built a cell-cell communication network using CellphoneDB and NicheNet. Cell-cell interactions indicated that ECs communicate extensively with one another. Interestingly, we discovered that intercellular communication between EC1 and EC2 was increased in Dupuytren's disease (Figs. 1O and Supplementary Fig. 1G). Significant signaling pathways including the MIF, CXCL14, CD44, CCL2, TNFSF12, and TNFSF10 pathways were identified in Dupuytren's disease interaction between EC1 and EC2, while JAG1, JAM2, and DLL1 pathways were found in healthy controls (Figs. 1O). The ligand-receptor pairs that may cause the transcriptome changes were predicted using NicheNet in order to further examine the cellular crosstalk between EC1 and EC2 in Dupuytren's disease. According to the results of NicheNet, MIF, and TNFSF12 were likewise included among the top anticipated ligands as EC2 as receiving cells (Figs. 1O). It's interesting to note that NicheNet analysis also predicted the genes that the ligand-receptor interactions would target. LINC01235 was among MIF's target genes for Dupuytren's disease (Fig. 1P). The findings highlighted the importance of interactions between EC1 ligands and EC2 receptors in controlling gene expression in Dupuytren's disease.
Overall, we uncovered molecular properties of ECs that correlate with Dupuytren's disease at the single-cell level, which extends our understanding of similar pathologic disorders. These findings could contribute to our understanding of the pathogenesis of Dupuytren's disease.
{kind=link}