Abundance of Prophages in NTM Species
Prophages were predicted in 81 of the 235 clinical NTM genomes (37.7% of complete genomes and 33.5% of draft genomes). Within the 81 genomes with prophages, a total of 96 unique prophages were identified. The interquartile range of prophage lengths across all species was ~38 kilobases to ~58 kilobases. A Kruskal-Wallis test of prophage lengths by growth rate failed to reject the null hypothesis suggesting no difference in the size of integrated prophages by growth rate (H=1.93, p=0.165). We predicted integrated prophage elements in both RGM and SGM, however predicted prophages more likely to be found within RGM than in SGM (F = 6.71, p = 1.96e-11 draft genomes and F = 3.83, p = 1.29e-04 complete genomes, proportions z-test , Fig. 1). In the RGM M. abscessus subsp. abscessus, 61 out of 109 (56.0%) clinical NTM have predicted prophage elements, while of the SGM, only 2 out of 63 M. avium (3.2%), 3 out of 23 M. intracellulare (13%) and 1 out of 11 M. chimaera (9.1%) draft genomes have intact prophage elements. The relative genomic locations of the predicted prophages within the complete genome dataset are shown in Supplementary Figure 1.
To test if the prophage discovery tools are biasing prophage predictions in longer contiguous sequences (i.e. complete genomes), we quantified the number of edge cases by species in the 182 draft genomes. The number of edge cases are normalized by genome count per species to generate an average number of edge cases per species (Table 1). Edge cases are only quantified in the draft genome assemblies because the complete genomes are assumed to have only one contiguous sequence (Table 2). Linear mixed models of prophage frequency by number of contigs (Z=-3.27, p=1.1e-03), N50 Length (Z=1.94, p=0.052), and number of contigs > 1500 base pairs (Z=-4.10, p=4.19e-04) approached or achieved significance. However, additional post-hoc testing of significant linear mixed models revealed no significant correlations suggesting assembly fragmentation likely had little effect on the number of predicted prophages between species.
Thus, in future figures, draft and complete genomes are combined for downstream analysis.
Table 1: Summary statistics of prophages predicted in the NTM draft genomes assemblies. Median counts of tRNA, N50 Lengths, and counts of contigs in NTM draft genomes are shown with the ranges in parenthesis. The edge case average is the total number of edge cases divided by the draft genome count.
|
Species
|
Genomes Count
|
Genomes with Phage
|
Predicted Prophages
|
Median tRNA Count
|
Median N50 Length
|
Median
Contig
Count
|
Edge Cases Average
|
|
M. avium
|
43
|
1
|
1
|
57 (54 – 108)
|
81670 (56262 – 116826)
|
206 (126 – 336)
|
1.07
|
|
M. chimera
|
11
|
1
|
1
|
55 (49 – 81)
|
86644 (78123 – 101816)
|
193 (116 – 251)
|
1.00
|
|
M. intracellulare
|
23
|
3
|
4
|
52 (50 – 54)
|
101938 (82877 – 131830)
|
104 (83 – 145)
|
0.13
|
|
M. abscessus
|
76
|
42
|
51
|
48 (45 – 116)
|
168941 (87141 – 327102)
|
67 (41 – 191)
|
2.60
|
|
M. bolletti
|
4
|
3
|
3
|
48.5 (48 – 49)
|
208710 (140476 – 226296)
|
45 (37 – 59)
|
2.50
|
|
M. massiliense
|
25
|
11
|
12
|
51 (46 – 84)
|
215865 (97542– 413240)
|
53 (39 – 143)
|
2.84
|
Table 2: Summary statistics of predicted prophages within complete NTM genomes selected from the NCBI assembly database. Complete genomes are not fragmented into contigs, thus N50 statistics, contig count, and edge cases are not applicable. Additional information about these genomes are available in Supplementary File 1.
|
Species
|
Genomes Count
|
Genomes with Phage
|
Predicted Prophages
|
Median tRNA Count
|
|
M. avium
|
20
|
1
|
1
|
59 (54 – 59)
|
|
M. abscessus
|
33
|
19
|
23
|
49 (46 – 104)
|
Predicted Prophages Annotated Using Existing Mycobacteriophage Clusters
PhagesDB is a database containing mycobacteriophage genomes categorized into clusters and sub clusters based on sequence similarity and shared functional genes. Predicted prophages in our study were annotated across 15 clusters (Fig. 2) using ANI and protein sequence similarity. The RGM had prophages in all 15 lettered groups, and SGM had 3 lettered groups. Most prophages (31.3%) fell into the “no cluster” category, which represents prophages matching other prophages without a defined cluster or not being assigned a cluster using ANI or BLASTp. The number of errors associated with the ANI assignment to lytic clusters is on average nearly double that of prophages assigned to lysogenic clusters (22,171 lytic against 12,385 lysogenic base pair errors, p=0.098, Mann Whitney U).
Across the entire dataset (draft and complete genomes), the 96 identified prophages consisted of 84 genotypically unique sequences. Predicted prophages that clustered with another predicted prophage were from the same species, which could represent a shared evolutionary sequence or a common insertion site.
The pangenome analysis using all predicted prophages resulted in no core genes shared among 96 unique prophages. The largest number of prophages that shared a gene was 25 prophages and the gene function was undefined. Supplementary Table 1 details the pangenome analyses, including shell gene counts between prophages discretized by assigned PhagesDB cluster. No core genes are found in any lettered cluster pangenome analysis. Hypothetical proteins of unknown function are most commonly shared shell genes within the predicted prophages.
Prophage Annotation Results and Virulence Genes by Species
There was an average of 68 open reading frames (ORFs) predicted within each of the 96 prophages from the draft and complete genomes. The average peptide length of ORFs was 216 (24 - 1937) amino acids. The total number of ORFs among all prophages was 6,550. Of these predicted ORFs, 65.24% were labeled hypothetical proteins without a known function. Almost half of the ORFs (48.89%) were annotated using sequence similarity to a protein within the PhagesDB mycobacteriophage database. ARAGORN identified 156 tRNAs with tRNA-threonine as the most abundant, comprising 16.0% of the total. The number of ORFs annotated as bacterial genes in the dataset, not including tRNAs, was 245 (3.74%). Of ORFs identified as bacterial genes, 66 ORFs (1.01% of all ORFs) were predicted to derive from bacterial virulence genes.
Virulence gene annotations within the predicted prophages comprised about 1% of the ORFs. The 66 predicted ORFs annotated as bacterial virulence genes were present across 39 prophages from 37 different NTM genomes. Figure 3 highlights the presence/absence of bacterial virulence gene within the predicted prophages across mycobacterial species. Of the genes annotated as virulence genes by VFDB or Patric-VF, 51.5% were originally identified in Mycobacteriumtuberculosis and 16.7% derive from Salmonella enterica (Supplementary File 2).
PhagesDB contains 1,795 mycobacteriophages. Within these mycobacteriophages, 187 mycobacteriophages contained 249 virulence genes or 0.13% of all predicted ORFs. For a fairer comparison of virulence gene abundance between our predicted prophages and PhagesDB, lysogenic mycobacteriophages were subset from PhagesDB leaving 1,271 mycobacteriophages and 122,405 ORFs. Within these 1,271 mycobacteriophages, 158 mycobacteriophages contain 220 annotated virulence genes (0.18% of all ORFs). We found that bacterial virulence genes are more likely to be present within prophage elements derived from clinical sources than from all mycobacteriophage genomes in PhagesDB isolated from the environmental M. smegmatis and the subset selected from lysogenic mycobacteriophages (F = 8.89, p = 6.16e-19 and F = 6.73, p = 1.73e-11, proportions z-test, Figure 4). The relative locations and functional annotations of bacterial virulence genes in the predicted prophages, as well as PhagesDB mycobacteriophages are shown in Figure 5. The location of the virulence genes in the predicted prophages, with 43.9% within 10% of either end of the predicted prophage, suggests either specialized transduction, where genes flanking the prophage insertion site are transduced or an error in the predicted prophage range (Fig. 5). 125 bacterial virulence genes (50.2%) are within 10% of mycobacteriophage ends in the PhagesDB database (Fig. 5). The presence of virulence genes in clinical isolates is significantly enriched even when these virulence genes near the ends are removed from only the predicted prophages (F = 5.03, p = 4.85e-7 and F = 3.37, p = 7.44e-4, proportions z-test).
Mycobacterium Phylogeny Influences on Prophage Frequency
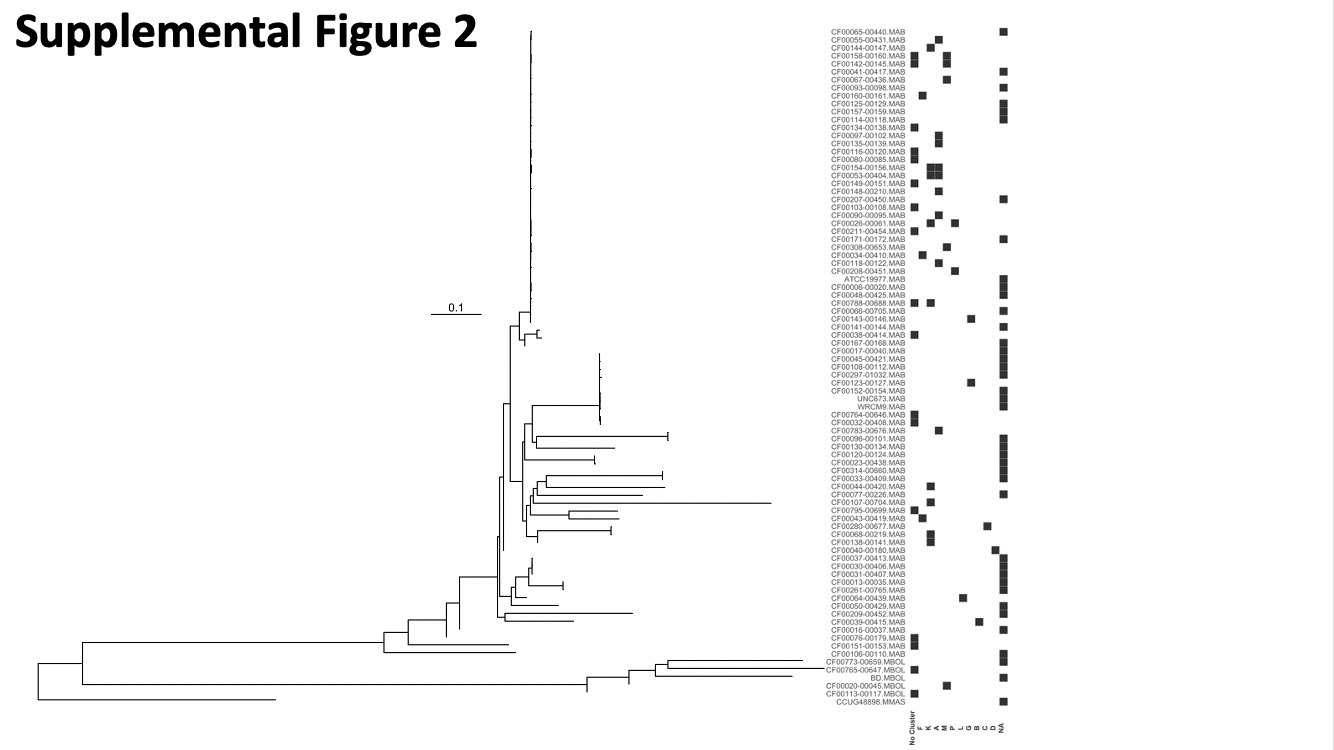
To test if NTM draft genomes with prophages are more evolutionarily similar to each other than to NTM draft genomes without prophage, we performed a PERMANOVA test of phylogenetic distance metrics. The genome wide genetic distances of M.abscessus subsp. abscessus draft genomes are more similar within the groups: with and without prophages, than between groups (F = 2.17, p = 0.026, PERMANOVA). Supplementary Figure 2 displays the distribution of prophages in M. abscessus subsp. abscessus and M. abscessus subsp. bolletti in the context of the bacterial phylogeny. The distributions of predicted prophages in other species are shown in Supplementary Figures 3 and 4.
CRISPR elements have the capability to protect a bacterium against prophage integration. CRISPR elements were present in only 4 of the 53 (7.5%) complete genomes and 12 of the 182 (6.6%) draft genomes. Presence of CRISPR elements was not associated with the number of prophages (H=0.0092, p=0.92, Kruskal-Wallis). The abundance of tRNAs in the mycobacterial genomes was significantly different by growth rate with slowly growing mycobacterial species having a greater number of tRNAs (H=89.43, p=3.18e-21, Kruskal-Wallis). In addition, the relationship of tRNAs and prophage frequency corrected by species reveals a positive linear correlation only in M. abscessus (R2=0.34, p=3.21e-4).
{kind=link}
{kind=link}
{kind=link}
{kind=link}