The synthesis and crystal structure of 2-bromo-4,6-bis(dibromoacetyl)resorcinol, I, was reported. In the title compound, I, crystalized in the triclinic crystal system with Pī space group. The two carbon and the oxygen atom of the acetyl groups (atoms C7, C8, O3, and C9, C10, O4) are nearly co-planar with the central phenyl ring. Intramolecular O–H···O, C–H···Br, and intermolecular C–H···Br/O interactions, two non-bonded contacts (Br5···Br3 and O4···C8) and π-π stacking interaction are stabilized the crystal packing of the title compound. Intermolecular interactions that exist in the title compound, I, are quantified with the aid of PIXEL and Hirshfeld surface (HS) analysis and the decomposed fingerprint (FP) plots. The FP plot reveals that the Br···Br contacts are comparably higher than the other contacts in the title crystal structure. Furthermore, the theoretical density functional theory (DFT) calculations were performed at the M062X /cc-PVTZ level of theory. The experimental geometry parameters of the title molecule are compared with the geometry of the optimized molecule in the gas phase. The chemical reactivity and charge transfer properties of the title compound were calculated from the HOMO and LUMO energy. In addition, the molecular electrostatic potential map was generated at their crystal structure geometry and quantitatively analyzed.
Research Article
Synthesis, Crystal structure, Hirshfeld surface analysis and DFT studies of 2-bromo-4,6-bis(dibromoacetyl)resorcinol
https://doi.org/10.21203/rs.3.rs-3086971/v1
This work is licensed under a CC BY 4.0 License
Journal Publication
published 19 Aug, 2023
You are reading this latest preprint version
Crystal structure
Intermolecular interactions
Hirshfeld Surface analysis
PIXEL
DFT
4, 6-Diacetylresorcinol and its derivatives have had great attention in the last few decades in the scientific community due to its utility key starting material for the synthesis of many organic and hybrid organic-inorganic materials, which has been used in heterocyclic, analytical, and pharmaceutical chemistry (Ali et al. 2013; Abdel-Rahman et al. 2015). Many researchers have synthesized the heterocyclic compounds and metal complexes from 4, 6-diacetylresorcinol, notably pharmacologically active molecules. and also explored their pharmacological activity(Ashok and Shravani 2009; Salavati-Niasari et al. 2009; Emara, El-Sayed, and Ahmed 2008; Krishna Murthy et al. 2002; Makode, Bhadange, and Aswar 2003). The structural features of 4, 6-diacetylresorcinol, particularly the mutual ortho position of acetyl and phenolic (-OH) group are favored to obtain oxygen- and nitrogen- containing heterocyclic systems as well as symmetric and asymmetric heterocyclic compounds(Abdel-Rahman et al. 2015; Ali et al. 2013).
The present work is a continuation of our earlier research work on the synthesis of coumarin, aminocyanopyridine and azo derivatives(Loganathan et al. 2015; Purushothaman, Loganathan, and Sithick 2012; Purushothaman et al. 2014). As a part of earlier studies, the title compound, 2-bromo-4,6-bis(dibromoacetyl)resorcinol (I) was synthesized from 4, 6-diacetylresorcinol and bromine in acetic acid. Herein, synthesis and the crystal structure of the title compound are reported. Based on the earlier studies, the various intermolecular interactions or nonbonded contacts that contribute toward crystal packing can be quantified by the PIXEL method and DFT calculation (Gavezzotti 2002, 2003; Gavezzotti 2011; Dey et al. 2014; Kaur et al. 2012; Khan et al. 2016; Panini, Bhandary, and Chopra 2016; Shukla and Chopra 2015; Panini and Chopra 2013; Shukla et al. 2017; Panini et al. 2013) and those contacts in the crystal structure visualized using the Hirshfeld surface[HS] analysis(Spackman and Jayatilaka 2009; McKinnon, Spackman, and Mitchell 2004; Parkin et al. 2007; Spackman and McKinnon 2002; Spackman, McKinnon, and Jayatilaka 2008; Turner et al. 2011; Wood et al. 2008) with the aid of 2D fingerprint plots(Spackman and McKinnon 2002; Kathiravan et al. 2016a, 2016b; Percino et al. 2017; Udayakumar et al. 2017; Venkatesan, Rajakannan, et al. 2016; Venkatesan et al. 2015; Venkatesan, Thamotharan, et al. 2016). Furthermore, the optimized molecular structure and HOMO-LUMO energies were calculated by the M062X/cc-PVTZ level of theory.
Chemicals
All the chemicals were purchased from Sigma-Aldrich chemical company and used without further purification.
Synthesis of 2-bromo-4,6-bis(dibromoacetyl)resorcinol ( I )
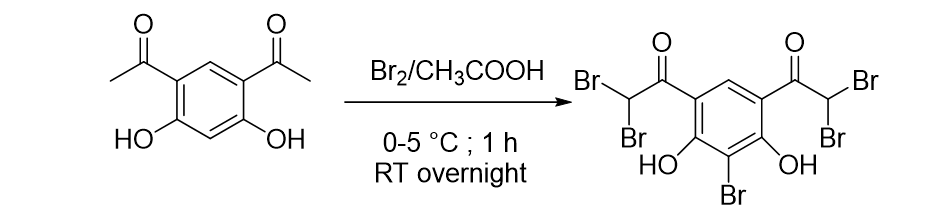
The title compound, 2-bromo-4,6-bis(dibromoacetyl)resorcinol, (I, Scheme 1), was synthesized from 4, 6-diacetylresorcinol (1.94 g, 10 mmol) and bromine (2.56 g, 50 mmol) in acetic acid (50 mL) at ice cold condition (0–5°C) for about one hour. The reaction content was kept overnight at room temperature (25°C ). The solid formed was filtered through the suction pump and dried in air. The solid crystallized from methanol gave orange colour crystals. m.p. 294°C.
X-Ray structure analysis and refinement
Single crystals of I were grown by slow evaporation method at room temperature from methanol. The X-ray intensity data were collected at room temperature (296 K) on a Bruker SMART APEX II CCD diffractometer (Data collection: APEX2; cell refinement SAINT; data reduction: SAINT and absorption correction: SADABS. Bruker AXS Inc., Madison, Wisconsin, USA) using Mo Kα radiation (λ = 0.71073 Å)(Clegg and Watson 2008). The structure was solved by the direct methods using the SHELXS-2014 program (Sheldrick 2015). All non-hydrogen atoms in I were refined by full-matrix least-squares on F2 using SHELXL-2018/3 (Sheldrick 2015). The positions of H atoms attached to oxygen atoms (O–H) were located from a different Fourier map and refined freely along with their isotropic displacement parameters. All the remaining H atoms were placed at the calculated positions (C–Haromatic and C–Hvinylic is 0.93 Å; and other C–H = 0.96–0.97 Å) using a riding model approach. The ORTEP and crystal packing diagrams were generated using the PLATON (Spek 2009), and Mercury (Macrae et al. 2006), respectively.
Computation calculation
All the quantum chemical calculations were performed using the Gaussian16, Revision B.01 [39]. The gas-phase structural optimization was carried out to compare the experimental geometry of the title compound (I). The X-ray geometry of title compound (I) was used as an initial model for the optimization calculation with the M06-2X/cc-pVTZ level of theory [40, 41] along with the incorporation of Grimme’s D3 dispersion correction [42]. Furthermore, the vibrational frequencies were calculated for the optimized molecule to confirm the proper convergence to energy minima on the potential energy surface (PES). The optimized molecular structure was used for further computations.
Hirshfeld surface (HS) analysis, PIXEL energy calculation, and molecular electrostatic potential calculations
The Hirshfeld surface (HS) and the decomposed 2D fingerprint (FP) plots were used to visually analyze the various intermolecular interactions in I. The HS and FP plots were generated with the aid of the CrystalExplorer17 program (Turner et al. 2017). Lattice energies and the intermolecular interaction energies for molecular pairs in the crystal structures of I were calculated by the CLP package (version 12.5.2014) with the PIXELC module (Gavezzotti 2008). The electron density for the title molecule has been calculated at the MP2/6-31G** level of theory with the aid of Gaussian16, Revision B.01 program and it was used for the PIXEL calculation. Overall lattice energies and interaction energies for various dimers partitioned into their Coulombic (Ecou), polarization (Epol), dispersion (Edisp), and repulsion (Erep) contributions terms were calculated. The percentage contributions of electrostatic and dispersion energies were computed as mentioned in our studies (Udayakumar et al. 2019; Venkatesan et al. 2019; Venkatesan et al. 2018). The molecular electrostatic potential map of I was generated at their crystal structure geometry from the corresponding wave function (which was calculated by Gaussian16, Revision B.01 with M06-2X/cc-pVTZ level of theory) using the WFA-SAS program (Bulat et al. 2010).
Structure description of I
The title compound, I is crystalized in the triclinic crystal system with the Pī space group. A single molecule is present in the asymmetric unit of I. The ORTEP view of the asymmetric unit of I with an atom numbering scheme is illustrated in Fig. 1. The crystal data and crystallographic refinement statistics are summarized in Table 1. The two carbon and the oxygen atoms of the acetyl groups (atoms C7, C8, O3, and C9, C10, O4) are nearly co-planar with the central phenyl ring in I. This is witnessed by the dihedral angle between the central phenyl ring and each of the two mean planes of acetyl groups is 3.65 (5)ο and 8.36 (3)ο.
| Parameters | I |
|---|---|
| CCDC number | 2260315 |
| Empirical formula | C10 H5 Br5 O4 |
| Formula weight | 588.69 |
| Wavelength (Ǻ) | 0.71073 |
| Crystal system and Space group | Triclinic and Pī |
| a (Ǻ) | 9.3626(2) |
| b (Ǻ) | 30.7365(7) |
| c (Ǻ) | 6.9825(1) |
| α (°) | 85.495 (5) |
| β (°) | 89.306(5) |
| γ (°) | 68.859(4) |
| V (Ǻ3) | 713.7(3) |
| Z | 2 |
| Calculated density (Mg/m3) | 1.394 |
| Absorption (mm− 1) | 14.08 |
| F(0 0 0) | 544 |
| Crystal size (mm) | 0.247× 0.223 ×0.161 |
| θ (°) | 1.8–30.1 |
| Limiting indices, h | −10≤ h ≥ 10 |
| k | −13 ≤ k ≥ 13 |
| l | −15 ≤ l ≥ 15 |
| Reflections collected/unique (Rint) | 148050/4127 |
| (θ °) Completeness (%) | 25.42, 99.9 |
| Refinement method | full-matrix least-squares on F2 |
| Data/restraints/parameters | 4127/0/176 |
| Goodness-of-fit (GOF) onF2 | 1.039 |
| Final R indices [I > 2σ(I)] | R1 = 0.0400 |
| wR2 = 0.0944 | |
| R indices (all data) | R1 = 0.0629 |
| wR2 = 0.1031 | |
| Largest difference in peak and hole (e A− 3) | 1.232 and − 1.053 |
To compare the geometric parameters of I with related crystal structure reported in Cambridge Structural Database (CSD, Version 5.43, last update Mar 2022)(Groom et al. 2016), a search was made with the 4, 6-diacetylresorcinol skeleton in the CSD using Conquest module (Bruno et al. 2002). It gave 18 hits along with two polymorphic forms of 4, 6-diacetylresorcinol (CSD reference code: VOXPED and VOXPED01). Further, to understand the geometrical difference in the title compound I, the geometric parameters of two polymorphs (VOXPED and VOXPED01) are compared with I. There is a small deviation in geometric parameters seemed between I and two polymorphs (VOXPED and VOXPED01). Particularly, the Ccarbonyl–Cmethyl bond length (C7–C8:1.524 (6); C9–C10:1.523 (6) Å) is slightly longer in I when compared with two polymorphs and it falls at 1.461(VOXPED), 1.469 (VOXPED01)Å, respectively. On the other hand, the bond lengths of Caromatic–Ccarbonyl and C = O in acetyl groups in I and two polymorphs (VOXPED and VOXPED01) are comparable with each other. This slight bond length change in the Ccarbonyl–Cmethyl bond might be due to steric hindrance and the electronic effect of two substituted bromine atoms in the acetyl group. Furthermore, the structural optimization calculation was performed in the gas phase using the X-ray geometry as an initial model. The M06-2X/cc-pVTZ level of theory [40, 41] along with the incorporation of Grimme’s D3 dispersion correction [42] was used. Vibrational frequencies were computed for the optimized structures to confirm the proper convergence to energy minima on their respective potential energy surfaces. No imaginary frequency was observed for the optimized structure.
| Atoms | Bond length, Å | Atoms | Bond angle, θ | ||
|---|---|---|---|---|---|
| X-ray | DFT | X-ray | DFT | ||
| Br1—C1 | 1.884(4) | 1.875 | C5—C9—C10 | 119.2 (3) | 118.2 |
| Br2—C10 | 1.917(5) | 1.917 | C9—C10—Br2 | 118.8 (3) | 111.8 |
| Br3—C10 | 1.933(5) | 1.944 | C9—C10—Br3 | 108.2 (3) | 107.0 |
| Br4—C8 | 1.928(5) | 1.934 | Br2—C10—Br3 | 110.5 (2) | 112.2 |
| Br5—C8 | 1.931(6) | 1.939 | Atoms | Torsion angle, θ | |
| O1—C6 | 1.335(5) | 1.318 | X-ray | DFT | |
| O2—C2 | 1.334(6) | 1.319 | C6—C1—C2—O2 | -179.6 (3) | 179.8 |
| O3—C7 | 1.231(6) | 1.221 | Br1—C1—C2—O2 | 1.7 (5) | -0.2 |
| O4—C9 | 1.224(4) | 1.215 | C6—C1—C2—C3 | 0.6 (6) | -0.4 |
| C1—C6 | 1.384(5) | 1.392 | Br1—C1—C2—C3 | -178.2 (3) | 179.7 |
| C1—C2 | 1.377(6) | 1.393 | O2—C2—C3—C4 | 179.8 (3) | -179.8 |
| C2—C3 | 1.434(6) | 1.426 | C1—C2—C3—C4 | -0.4 (5) | 0.4 |
| C3—C4 | 1.396(6) | 1.387 | O2—C2—C3—C7 | -2.0 (5) | -0.4 |
| C3—C7 | 1.455(5) | 1.460 | C1—C2—C3—C7 | 177.9 (3) | 179.8 |
| C4—C5 | 1.384(5) | 1.384 | C2—C3—C4—C5 | -0.2 (5) | 0.5 |
| C5—C6 | 1.432(6) | 1.427 | C7—C3—C4—C5 | -178.4 (3) | -178.9 |
| C5—C9 | 1.456(6) | 1.467 | C4—C5—C6—C1 | -0.4 (5) | 1.3 |
| C7—C8 | 1.524(6) | 1.530 | C9—C5—C6—C1 | -179.4 (3) | -177.9 |
| C9—C10 | 1.523(6) | 1.532 | C4—C3—C7—O3 | 179.3 (4) | 179.6 |
| Atoms | Bond angle, θ | C2—C3—C7—O3 | 1.2 (6) | 0.2 | |
| X-ray | DFT | C4—C3—C7—C8 | 1.5 (6) | -0.5 | |
| C6—C1—C2 | 122.3 (4) | 121.0 | C2—C3—C7—C8 | -176.7 (4) | -179.9 |
| C6—C1—Br1 | 118.9 (3) | 119.5 | O3—C7—C8—Br4 | 121.6 (3) | -116.9 |
| C2—C1—Br1 | 118.8(4) | 119.5 | C3—C7—C8—Br4 | -60.4 (5) | -65.6 |
| O2—C2—C1 | 118.7 (4) | 118.4 | C3—C7—C8—Br5 | 66.7 (4) | -65.6 |
| O2—C2—C3 | 121.7 (4) | 122.0 | C3—C4—C5—C6 | 0.6 (5) | -1.4 |
| C1—C2—C3 | 119.6 (4) | 119.7 | C3—C4—C5—C9 | 179.6 (3) | 177.9 |
| C4—C3—C2 | 117.6 (4) | 118.6 | C2—C1—C6—O1 | -178.4 (3) | 179.8 |
| C4—C3—C7 | 123.3 (4) | 122.6 | Br1—C1—C6—O1 | 0.4 (5) | -0.2 |
| C2—C3—C7 | 119.0(4) | 118.8 | C2—C1—C6—C5 | -0.2 (5) | -0.4 |
| C5—C4—C3 | 122.8 (4) | 122.5 | Br1—C1—C6—C5 | 178.6 (3) | 179.5 |
| C4—C5—C6 | 118.7 (4) | 118.7 | C4—C5—C6—O1 | 177.7 (3) | -178.9 |
| C4—C5—C9 | 122.6 (3) | 122.4 | C9—C5—C6—O1 | -1.3 (5) | 1.8 |
| C6—C5—C9 | 118.7 (4) | 118.8 | C4—C5—C9—O4 | -171.4 (4) | 177.6 |
| O1—C6—C1 | 118.8 (4) | 118.6 | C6—C5—C9—O4 | 7.5 (5) | -3.2 |
| O1—C6—C5 | 122.1 (4) | 121.8 | C4—C5—C9—C10 | 7.5 (5) | -2.7 |
| C1—C6—C5 | 119.1 (4) | 119.6 | C6—C5—C9—C10 | -171.8 (3) | 176.6 |
| O3—C7—C3 | 122.3 (4) | 122.7 | O4—C9—C10—Br2 | 13.1 (5) | 24.38 |
| O3—C7—C8 | 114.3 (4) | 114.5 | C5—C9—C10—Br2 | -167.6 (3) | -155.4 |
| C3—C7—C8 | 123.3 (4) | 122.8 | O4—C9—C10—Br3 | -108.8 (4) | -98.8 |
| C7—C8—Br4 | 113.7 (3) | 112.4 | C5—C9—C10—Br3 | -108.8 (4) | 81.5 |
| C7—C8—Br5 | 110.7 (2) | 111.5 | C6—C1—C2—O2 | -179.6 (3) | 179.8 |
| Br4—C8—Br5 | 112.1 (2) | 113.5 | Br1—C1—C2—O2 | 1.7 (5) | -0.2 |
| O4—C9—C5 | 122.2 (4) | 122.4 | C6—C1—C2—C3 | 0.6 (6) | -0.4 |
| O4—C9—C10 | 118.7(4) | 119.4 | |||
As seen in Table 2, the bond lengths, bond angles, and torsion angles of experimental values and theoretical values are comparable with each other. The structural overlay diagram of the crystal structure and their optimized molecule is shown in Fig. 2. The atoms C1–C7, Br1, and C9 are used to overlay. Both structures are well overlaid with each other with a root mean square deviation (r.m.s.d) is 0.0177 Å except for bromine atoms in the acetyl group. The deviation around bromine atoms is witnessed by the largest deviations of bond lengths of C10–Br3 and C1–Br1 (0.01 Å) and bond angles of Br4–C8–Br5 (1.4°) and Br2–C10–Br3 (1.7°, Table 2). Similarly, the largest deviations of torsion angle ≈ 10–20° were observed around the bromine atoms in experimental and optimized molecules (Table 2). These deviations can be attributed to the fact that the theoretical calculations have been carried out with an isolated molecule in the gaseous phase and the experimental values correspond to the molecule in the crystalline state.
Crystal packing, Molecular dimers, Intermolecular interactions energy, and Lattice energy of I
The crystal packing of the title compound is shown in Fig. 3. As can be seen from Fig. 3, molecules are arranged in a column manner. The crystal packing of I is stabilized by the intramolecular O–H···O, C–H···Br interactions. Two intramolecular O–H···O (O2–H2···O3 and O1–H1···O4) interactions individually makes pesudo S(6) ring. The total lattice energy for the title compound is stabilized by −125.9 kcal mol−1.
Apart from intramolecular interaction, the four types of intermolecular interactions (C–H···Br, C–H···O, Cg···Cg, and Br···Br) play a vital role in the stabilization of crystal packing of I. The energetically significant five molecular dimers/pairs (M1-M5) were identified from the crystal packing of I with the aid of PIXEL analysis(Fig. 4.). These dimers are listed in Table 3 with the decreasing order of their stabilization energy (Etot). The Etot of these dimers falls in the range of Etot: -13.1 – -1.8 kcal mol-1.
| Dimers | Important interactions | Geometry (Ȧ, °) | Symmetry operator | Distance (Å) | Ecoul | Epol | Edis | Erep | Etot | % Edis | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| d(H···A) | d(D···A) | <D-H···A | ||||||||||
| M1 | Cg1···Cg1 | 3.933 | -x + 2, -y, -z + 1 | 4.791 | -6.5 | -3.0 | -18.8 | 15.2 | -13.1 | 66 | ||
| C10-H10···Br1 | 3.04 | 3.605 | 119 | |||||||||
| M2 | Cg1···Cg1 | 3.283 | -x + 2, -y + 1, -z + 1 | 6.635 | -3.1 | -0.8 | -10.0 | 5.8 | -8.0 | 72 | ||
| M3 | C10-H10···Br2 | 2.92 | 3.664 | 134 | -x + 2, -y, -z | 9.116 | -4.0 | -1.1 | -9.7 | 8.1 | -6.6 | 66 |
| M4 | C8-H8···O4 = C9 | 2.26 | 3.207 | 164 | x-1, y + 1, z | 9.645 | -4.4 | -1.3 | -4.9 | 6.0 | -4.6 | 46 |
| M5 | Br3···Br5 | 3.671 | x-1, y, z | 7.306 | -0.4 | -0.3 | -2.9 | 1.7 | -1.8 | 81 | ||
| Total lattice energy | -61.3 | -19.1 | -173.5 | 128.1 | -125.9 | |||||||
Among the five molecular pairs, the strongest molecular pair, M1 is stabilized (Etot: -13.1 kcal mol-1) by π ···π stacking interaction. Briefly, the two phenyl rings in M1 are arranged in a parallel-displaced manner(Venkatesan et al. 2018; Riwar et al. 2017) with the distance between both centroids of phenyl rings is 3.934 (3) Å (slippage: 1.984 Å, symmetry code: 2-x, -y, 1-z). The dihedral angle between both phenyl ring planes is 0° which indicates that both phenyl rings are in a parallel manner. This dimer, M1, is further stabilized by the C10-H10···Br1 interaction. The second strongest molecular dimer, M2 (Etot: -8.0 kcal mol-1) is also stabilized by another stacking interaction that formed between two pseudo rings with the distance between both centroids of two pseudo rings being 3.283 Å. Briefly, the intramolecular O2–H2···O3 interaction makes S(6) ring motif as mentioned earlier. In this S(6) ring motif stacked with another S(6) ring of the neighbouring molecule. Similar kinds of stacking interactions were observed in geminal amido-esters(Venkatesan et al. 2021b, 2021a) and other compounds(Blagojević and Zarić 2015). Apart from these two stacking pairs, the molecular pairs M3 (Etot: -6.6 kcal mol-1) and M4 (Etot: -4.6 kcal mol-1) are stabilized by C10–H10···Br2 and C8–H8···O4 = C9 interaction respectively. The C8–H8···O4 = C9 interaction in M4 links the neighbouring molecules into C8 motif and C10–H10···Br2 interaction in M3 links neigbhouring molecules into centrosymmetric dimeric structure with \({R}_{2}^{2}\left(6\right)\) motif. The combination of interactions in M3 and M4 dimers links neigbhouring molecules into the molecular layer which extends parallel to the 110 plane (Fig. 5).
In addition to C–H···Br/O and Cg···Cg interactions, a non-bonded Br5···Br3 contact in M5 with distance is 3.6706 (13) Å which additionally stabilized the crystal structure of I. The Etot for M5 is -1.8 kcal mol-1. It should be mentioned that the Br···Br contact is shorter (0.029 Å) than the sums of the van der Waals radii of two bromine atoms. The CSD search suggested that the 3232 hits were found for the Br···Br (Lieberman, Davey, and Newsham 2000) contacts. As can be seen from Table 2, the % dispersion contribution (% Edis=Edis/(EpolEdisp+Edis)) is higher in M5 (81%) and the least 46% is observed in M4. These results suggested that the Br5···Br3 contact is more dispersive in nature whereas the C8–H8···O4 = C9 interaction in M4 in the Coulombic (Ecou:56%) nature.
Hirshfeld Surface analysis
To understand the role of various intermolecular interactions in a qualitative manner, we employed Hirshfeld surface analysis(Spackman and Jayatilaka 2009; Spackman and McKinnon 2002; Turner et al. 2011; Wood et al. 2008; Spackman, McKinnon, and Jayatilaka 2008). The Hirshfeld surface analysis and two-dimensional fingerprint plots were generated from the CrystalExplorer17(Turner et al. 2017). The HS and 2D-FP were used to provide additional information and to quantify the intermolecular interactions in the title compound by using distinct colours and intensities to indicate short and long contacts, as well as the relative contribution of the different interactions in their solid-state (Venkatesan, Thamotharan, et al. 2016; Venkatesan, Rajakannan, et al. 2016; Venkatesan et al. 2015). The Hirshfeld surfaces (HS) mapped over the dnorm (-0.0155 to 1.0144 a.u.) and shape index are shown in Fig. 6.
The red spots on the HS map are visible for the C10–H10···Br2(M3), C8–H8···O4(M4), and Br5···Br3 (M5) interactions. The π stacking interaction between two phenyl rings is confirmed by the red and blue triangles on the surface of the shape-index diagram (Venkatesan et al. 2018). The two-dimensional fingerprint plot (2D-FP) and decomposed plots are shown in Fig. 7. The 2D-FP plots showed that the Br···Br (35.8%), Br···H (18.9%), O···H (10.5%) and Br···O (9.8%) contacts are first four most significant contacts in their crystal structure. The relative contribution of H···H contact is 1.9% which is lower than the C···C contact (2.8%) in the crystal structure of I. It indicates the steric hindrance of bromine atoms in the title compound I.
Quantitative molecular electrostatic potential map (MESP)
The molecular electrostatic potential map of I was generated at their crystal structure geometry from the corresponding wave functions using the WFA-SAS program(Bulat et al. 2010). The molecular electrostatic potential maps of I along with the location of the positive (Vsmax, black hemisphere, black coloured values) and negative potentials (Vsmin, blue hemisphere, red coloured value) are shown in Fig. 8.
From Fig. 8, the positive potentials are observed on the surface of bromine atoms and hydrogen atoms in phenolic and acetyl group (dibromide substituted terminal methyl group in the acetyl moiety). Interestingly, the distribution of electrostatic potential is found to be asymmetric in nature in all the five bromine atoms in the I. The highest Vsmax: 34.8 kcal mol-1 in one of the bromine (Br2) atom in the diacetyl group whereas, another bromine (Br1) atom attached in the phenyl ring has the lowest Vsmax: 13.1 kcal mol-1. The positive potentials on the surface of bromine atoms suggested that the interactions involved in the bromine atoms in I belong in the σ-hole interaction. The highest negative potential (Vsmin) is -13.8 kcal mol-1 and it is observed on the surface of oxygen O4 of the acetyl group. These molecular electrostatic differences witness the involvement of these atoms in the C8–H8···O4, C10–H10···Br2 interactions in the crystal packing.
Frontier Molecular Orbital Analysis
It is well known that frontier molecular orbital analysis is one of the potential tools for the study of molecular electronic charge mobility, the chemical reactivity, kinetic stability of molecules, and electronic transitions in the molecules. To understand the intramolecular charge transfer (ICT) process in I, frontier molecular orbital analysis is carried out. As seen from Fig. 9, the highest occupied molecular orbital (HOMO) is mainly localized in the central phenyl ring, hydroxy group, and bromine atom(Br1) which are attached in the phenyl ring whereas the lowest unoccupied molecular orbital (LUMO) is localized in the dibromoacetyl moiety of the title compound. The HOMO and LUMO diagram shows the existence of the intramolecular charge transfer in the title compound. The HOMO→LUMO energy(EH-L) gap was found to be 6.292 eV and the frontier molecular orbital energies, EHOMO and ELUMO, were − 8.357 eV and − 2.065 eV, respectively.
To understand the chemical reactivity and stability of I, the DFT global parameters are calculated. In the present investigation, the ionization potential (IP), electron affinity (EA), chemical hardness (Ƞ), chemical potential (µ), and electronegativity (χ) are computed. The calculated ionization potential (IP, Eq. 1) and electron affinity can be used to predict the electronic stability of I. The IP and EA values are obtained by Koopmans approximations, the negative of HOMO is taken as IP whilst the negative of LUMO is EA (Eq. 1–2). The calculated ionization potential (IP) and electron affinity (EA) values are 8.357 eV and 2.065 eV, respectively. The significant IP and EA values suggest its better electronic stability and it has a strong tendency to attract the valence electrons, and the least electropositive nature.

The global hardness (Ƞ, Eq. 3) is defined by the first and second-order partial derivatives of total energy (E) with respect to the number of electrons (N) at constant external potential v(r), e

The chemical hardness(η, Eq. 4 ) determines how resistant a cluster of nuclei and electrons is to variations in the distribution of its electrons, and the calculated chemical hardness (η) for I is 3.146 eV. The calculated chemical potential (µ, Eq. 5 ) value of I is − 5.211 eV and this smaller value suggests that the title compound has a soft nature and it has reactive toward incoming reagents.

The electrophilicity index (ω) value indicates the amount of energy lost as a result of the maximum amount of electron movement in the molecule. The calculated electrophilicity index (ω, Eq. 7) value of I is 4.316 eV and indicates that the compound has a good electron mobility nature.
In conclusion, the synthesis and crystal structure of 2-bromo-4,6-bis(dibromoacetyl)resorcinol (I) are reported. The co-planarity of acetyl groups and central phenyl ring in I was revealed by the single crystal X-ray diffraction study. Two pseudo S(6) ring motifs were formed by the intramolecular O–H···O, C–H···Br interactions. The Hirshfeld surface analysis signaled that the intermolecular Br···Br, Br···H, O···H, and Br···O interactions seem to be important for the stabilization of I. Further, the role of various intermolecular interactions, C–H···Br, C–H···O, Cg···Cg and Br···Br in the stabilization of crystal packing of I was characterized by the PIXEL energy analysis. The energetically significant five molecular dimers/pairs (M1-M5), with a fall in the range of Etot: -13.1 – -1.8 kcal mol-1, were extracted from the crystal packing of I. The molecular electrostatic potential map of I was quantitatively analyzed and the positive potentials were observed on the surface of bromine atoms and hydrogen atoms in phenolic and acetyl groups. While the negative potential was observed on the surface of oxygen of the acetyl group. Furthermore, the electronic structure properties, such as HOMO→LUMO energy(EH-L) gap, the energy gap, the ionization potential, the electron affinity, the electronegativity, hardness, softness, chemical potential, and the electrophilicity index were calculated.
Acknowledgment
The authors PV and MJP wish to express their gratitude to VIEP-BUAP (grant pv19-ID00375, and grant 00110-VIEP-202) also the authors thankfully acknowledge the computer resources, technical and support provided by the Laboratorio Nacional de Supercómputo del Sureste de México. KL and MP wish to thank the Jamal Mohamed College management for providing the necessary facilities.
Funding
The authors PV and JP have received research support from VIEP-BUAP (grant pv19-ID00375, and grant 00110-VIEP-202).
Competing Interests
Financial interests: Authors PV and JP have research funding from VIEP-BUAP (grant pv19-ID00375, and grant 00110-VIEP-202) and other authors declare they have no financial interests.
Conflict of interest
The authors declare no conflict of interest.
Ethical Approval
Not applicable.
Authors' contributions
K. Loganathan: Conceptualization, Chemical synthesis, Characterization, A. Anandan: Conceptualization, Investigation, Crystal structure analysis and, Characterization, Writing – original draft, M. Purushothaman: Conceptualization, Chemical synthesis, Characterization, Writing- review & editing P. Daniel Jebaraj: Chemical synthesis, K. Thanigaimani: Data curation, review; M. Judith Percino: Resources, Data curation, Writing- review & editing P. Venkatesan: Conceptualization, Investigation, Supervision, Writing – original draft – review & editing.
Availability of data and materials
Data and materials will be made available on request.
- Abdel-Rahman, Reda M., Mounir A. I. Salem, Tarik E. Ali, and Magdy A. Ibrahim. 2015. 'Utility of 4,6-Diacetylresorcinol in Heterocyclic Synthesis',Chem Heterocycl Comp., 51: 299-309. https://doi.org/10.1007/s10593-015-1699-0.
- Ali, Tarik E., Magdy A. Ibrahim, Zeinab M. El-Gendy, and Eman M. El-Amin. 2013. '4,6-Diacetylresorcinol in Heterocyclic Synthesis, Part I: Synthesis and Biological Evaluation of Some New Linearly and Angularly Substituted Pyrano[3,2-g] Chromenes via Vilsmeier–Haack Formylation of 4,6-Diacetylresorcinol, Its Schiff Bases, and Hydrazones',Synth. Commun., 43: 3329-41. https://doi.org/10.1080/00397911.2013.783074.
- Ashok, D, and D Shravani. 2009. 'Microwave-Assisted Solvent Free Synthesis of Some 4, 6-Dicinnamoyl Resorcinols', Asian J. Chem. 808-10. https://asianjournalofchemistry.co.in/User/ViewFreeArticle.aspx?ArticleID=21_1_103
- Blagojević, Jelena P., and Snežana D. Zarić. 2015. 'Stacking interactions of hydrogen-bridged rings – stronger than the stacking of benzene molecules', Chem.Commun., 51: 12989-91. https://doi.org/10.1039/C5CC04139B
- Bruno, Ian J., Jason C. Cole, Paul R. Edgington, Magnus Kessler, Clare F. Macrae, Patrick McCabe, Jonathan Pearson, and Robin Taylor. 2002. 'New software for searching the Cambridge Structural Database and visualizing crystal structures', Acta. Crystallogr. B. Struct. Sci. Cryst. Eng. Mater., 58: 389-97. https://doi.org/10.1107/S0108768102003324
- Bulat, Felipe A., Alejandro Toro-Labbé, Tore Brinck, Jane S. Murray, and Peter Politzer. 2010. 'Quantitative analysis of molecular surfaces: areas, volumes, electrostatic potentials and average local ionization energies', J. Mol. Model., 16: 1679-91. DOI: 10.1007/s00894-010-0692-x
- Clegg, William, and David G. Watson. 2008. 'Structure Reports Online: major changes in response to a huge success', Acta Crystallogr. E: Crystallogr. Commun., 64: e15-e17. DOI: 10.1107/S1600536808002778
- Dey, Dhananjay, T. P. Mohan, B. Vishalakshi, and Deepak Chopra. 2014. 'Computational Study of the Formation of Short Centrosymmetric N–H···S Supramolecular Synthon and Related Weak Interactions in Crystalline 1,2,4-Triazoles', Cryst. Growth Des., 14: 5881-96. https://doi.org/10.1021/cg501103c
- Emara, Adel A. A., Badr A. El-Sayed, and El-Sayed A. E. Ahmed. 2008. 'Syntheses, spectroscopic characterization and thermal behavior on novel binuclear transition metal complexes of hydrazones derived from 4,6-diacetylresorcinol and oxalyldihydrazine', Spectrochim. Acta - A: Mol. Biomol. 69. 757-69. https://doi.org/10.1016/j.saa.2007.05.028
- Gavezzotti, A. 2002. 'Calculation of Intermolecular Interaction Energies by Direct Numerical Integration over Electron Densities. I. Electrostatic and Polarization Energies in Molecular Crystals', J. Phys. Chem. B., 106: 4145-54. https://doi.org/10.1021/jp0144202
- Gavezzotti, A. 2003. 'Calculation of Intermolecular Interaction Energies by Direct Numerical Integration over Electron Densities. 2. An Improved Polarization Model and the Evaluation of Dispersion and Repulsion Energies', J. Phys. Chem. B., 107: 2344-53. https://doi.org/10.1021/jp022288f.
- Gavezzotti, A. 2008. 'Non-conventional bonding between organic molecules. The ‘halogen bond’ in crystalline systems', Mol. Phys., 106: 1473-85. DOI: 10.1080/00268970802060674
- Gavezzotti, Angelo. 2011. 'Efficient computer modeling of organic materials. The atom-atom, Coulomb-London-Pauli (AA-CLP) model for intermolecular electrostatic-polarization, dispersion and repulsion energies', New J. Chem., 35: 1360-68. https://doi.org/10.1039/C0NJ00982B
- Groom, Colin R., Ian J. Bruno, Matthew P. Lightfoot, and Suzanna C. Ward. 2016. 'The Cambridge Structural Database', Acta. Crystallogr. B. Struct. Sci. Cryst. Eng. Mater., 72: 171-79. doi: 10.1107/S2052520616003954
- Kathiravan, Perumal, Thangavelu Balakrishnan, Perumal Venkatesan, Kandasamy Ramamurthi, Maria Judith Percino, and Subbiah Thamotharan. 2016a. 'Crystal structure and Hirshfeld surface analysis of 1-carboxy-2-(3,4-dihydroxyphenyl)ethan-1-aminium bromide 2-ammonio-3-(3,4-dihydroxyphenyl)propanoate', Acta Crystallogr. E: Crystallogr. Commun., 72: 1544-48. doi: 10.1107/S2056989016015425
- Kathiravan, Perumal, Thangavelu Balakrishnan, Perumal Venkatesan, Kandasamy Ramamurthi, Maria Judith Percino, and Subbiah Thamotharan. 2016b. 'Crystal structure and Hirshfeld surface analysis of 1-carboxy-2-(3,4-dihydroxyphenyl)ethan-1-aminium chloride 2-ammonio-3-(3,4-dihydroxyphenyl)propanoate: a new polymorph of l-dopa HCl and isotypic with its bromide counterpart', Acta Crystallogr. E: Crystallogr. Commun., 72: 1628-32. DOI: 10.1107/S2056989016016789
- Kaur, Gurpreet, Piyush Panini, Deepak Chopra, and Angshuman Roy Choudhury. 2012. 'Structural Investigation of Weak Intermolecular Interactions in Fluorine Substituted Isomeric N-Benzylideneanilines', Cryst. Growth Des., 12: 5096-110. https://doi.org/10.1021/cg3010294
- Khan, Imtiaz, Piyush Panini, Salah Ud-Din Khan, Usman Ali Rana, Hina Andleeb, Deepak Chopra, Shahid Hameed, and Jim Simpson. 2016. 'Exploiting the Role of Molecular Electrostatic Potential, Deformation Density, Topology, and Energetics in the Characterization of S···N and Cl···N Supramolecular Motifs in Crystalline Triazolothiadiazoles', Cryst. Growth Des., 16: 1371-86. https://doi.org/10.1021/acs.cgd.5b01499
- Krishna Murthy, K. S., B. Rajitha, M. Kanakalingeswara Rao, T. Raja Komuraiah, and S. M. Reddy. 2002. 'Facile Synthesis of Biologically Active Linear Bisaroyl Benzodifurans By PTC and Solvent Free Microwave Irradiation', Heterocycl Comm., 8: 179-86. https://doi.org/10.1515/HC.2002.8.2.179
- Lieberman, H. F., R. J. Davey, and D. M. T. Newsham. 2000. 'Br···Br and Br···H Interactions in Action: Polymorphism, Hopping, and Twinning in 1,2,4,5-Tetrabromobenzene', Chem. Mater., 12: 490-94. https://doi.org/10.1021/cm991123p.
- Loganathan, K, K Sithick Ali, M Purushothaman, S Silambarasan, and A Jamal Abdul Nasser. 2015. 'Synthesis and characterization of Azo derivatives of diacetylresorcinol', J Chem Pharm Res., 7: 1452-55. https://www.jocpr.com/abstract/synthesis-and-characterization-of-azo-derivatives-of-diacetylresorcinol-4725.html
- Macrae, Clare F., Paul R. Edgington, Patrick McCabe, Elna Pidcock, Greg P. Shields, Robin Taylor, Matthew Towler, and Jacco van de Streek. 2006. 'Mercury: visualization and analysis of crystal structures', J. Appl. Crystallogr. 39: 453-57. https://doi.org/10.1107/S002188980600731X
- Makode, J, JT Bhadange, and AS Aswar. 2003. 'Structural, thermal, biological and semiconducting properties of Mn (II), Fe (II), Co (II), Ni (II), Cu (II), Zn (II), Cd (II) and VO (IV) complexes of Schiff base derived from resdiacetophenone and S-benzyldithiocarbazate', Pol. J. Chem., 77: 855-65. https://www.infona.pl/resource/bwmeta1.element.baztech-article-BUJ1-0021-0046
- McKinnon, Joshua J., Mark A. Spackman, and Anthony S. Mitchell. 2004. 'Novel tools for visualizing and exploring intermolecular interactions in molecular crystals', Acta. Crystallogr. B. Struct. Sci. Cryst. Eng. Mater., 60: 627-68. doi: 10.1107/S0108768104020300
- Panini, Piyush, Subhrajyoti Bhandary, and Deepak Chopra. 2016. 'Quantitative Investigation of Polymorphism in 3-(Trifluoromethyl)-N-[2-(trifluoromethyl)phenyl]benzamide', Cryst. Growth Des., 16: 2561-72. https://doi.org/10.1021/acs.cgd.5b01638
- Panini, Piyush, and Deepak Chopra. 2013. 'Quantitative insights into energy contributions of intermolecular interactions in fluorine and trifluoromethyl substituted isomeric N-phenylacetamides and N-methylbenzamides', CrystEngComm, 15: 3711-33. https://doi.org/10.1039/C3CE40111A.
- Panini, Piyush, T. P. Mohan, Usma Gangwar, Ravish Sankolli, and Deepak Chopra. 2013. 'Quantitative crystal structure analysis of 1,3,4-thiadiazole derivatives', CrystEngComm, 15: 4549-64. https://doi.org/10.1039/C3CE40278A
- Parkin, Andrew, Gordon Barr, Wei Dong, Christopher J. Gilmore, Dylan Jayatilaka, Joshua J. McKinnon, Mark A. Spackman, and Chick C. Wilson. 2007. 'Comparing entire crystal structures: structural genetic fingerprinting', CrystEngComm, 9: 648-52. https://doi.org/10.1039/B704177B
- Percino, Judith, Margarita Cerón, Perumal Venkatesan, Paulina Ceballos, Alejandro Bañuelos, Oscar Rodríguez, Maxime A. Siegler, Fernando Robles, Víctor M. Chapela, Guillermo Soriano-Moro, Enrique Pérez-Gutiérrez, José Bonilla-Cruz, and Subbiah Thamotharan. 2017. 'Two Different Emissions of (2Z)-2-(4-Bromophenyl)-3-[4-(dimethylamino)phenyl]prop-2-enenitrile Due to Crystal Habit and Size: Synthesis, Optical, and Supramolecular Characterization', Cryst. Growth Des., 17(4) 1679-94. https://doi.org/10.1021/acs.cgd.6b01670
- Purushothaman, M, K Loganathan, KM Sithick Ali, and Joseph A Selvin. 2014. 'An efficient and facile synthesis of coumarin derivatives as potent antimicrobial agents', Int. J. Chem. Tech. Res, 6: 538-46. https://sphinxsai.com/2014/ChemTech/JM14CT51_100/CT=64(538-546)JM14.pdf
- Purushothaman, M, K Loganathan, and AK Sithick. 2012. 'Synthesis, characterization, and biological importance of aminocyanopyridines', Int J Chem Tech Res, 4: 479-83. https://sphinxsai.com/2012/chemAJ/CHEM/CT=02[479-483]AJ12.pdf
- Riwar, Leslie-Joana, Nils Trapp, Bernd Kuhn, and François Diederich. 2017. 'Substituent Effects in Parallel-Displaced π–π Stacking Interactions: Distance Matters', Angew. Chem. Int. Ed., 56: 11252-57. https://doi.org/10.1002/anie.201703744
- Salavati-Niasari, Masoud, Zohreh Salimi, Mahdi Bazarganipour, and Fatemeh Davar. 2009. 'Synthesis, characterization and catalytic oxidation of cyclohexane using a novel host (zeolite-Y)/guest (binuclear transition metal complexes) nanocomposite materials', Inorganica Chim. Acta., 362: 3715-24. https://doi.org/10.1016/j.ica.2009.04.028.
- Sheldrick, George. 2015. 'Crystal structure refinement with SHELXL', Acta Crystallogr. C Struct. Chem., 71: 3-8. https://doi.org/10.1107/S2053229614024218
- Shukla, Rahul, and Deepak Chopra. 2015. 'Crystallographic and computational investigation of intermolecular interactions involving organic fluorine with relevance to the hybridization of the carbon atom', CrystEngComm, 17: 3596-609. https://doi.org/10.1039/C4CE02391A
- Shukla, Rahul, Aamer Saeed, Jim Simpson, and Deepak Chopra. 2017. 'Quantitative investigation of C-H[three dots, centered][small pi] and other intermolecular interactions in a series of crystalline N-(substituted phenyl)-2-naphthamide derivatives', CrystEngComm, 19: 5473-91. https://doi.org/10.1039/C7CE01310H
- Spackman, Mark A., and Dylan Jayatilaka. 2009. 'Hirshfeld surface analysis', CrystEngComm, 11: 19-32. https://doi.org/10.1039/B818330A
- Spackman, Mark A., and Joshua J. McKinnon. 2002. 'Fingerprinting intermolecular interactions in molecular crystals', CrystEngComm, 4: 378-92. https://doi.org/10.1039/B203191B
- Spackman, Mark A., Joshua J. McKinnon, and Dylan Jayatilaka. 2008. 'Electrostatic potentials mapped on Hirshfeld surfaces provide direct insight into intermolecular interactions in crystals', CrystEngComm, 10: 377-88. https://doi.org/10.1039/B715227B
- Spek, Anthony. 2009. 'Structure validation in chemical crystallography', Acta Crystallographica Section D, 65: 148-55. https://doi.org/10.1107/S090744490804362X
- Turner, Michael J., Joshua J. McKinnon, Dylan Jayatilaka, and Mark A. Spackman. 2011. 'Visualisation and characterisation of voids in crystalline materials', CrystEngComm, 13: 1804-13. https://doi.org/10.1039/C0CE00683A
- Turner, MJ, JJ McKinnon, SK Wolff, DJ Grimwood, PR Spackman, D Jayatilaka, and MA Spackman. 2017. 'CrystalExplorer17', University of Western Australia.
- Udayakumar, M., M. Cerón, P. Ceballos, M. J. Percino, and S. Thamotharan. 2019. 'Interplay of weak noncovalent interactions in two conjugated positional isomers: A combined X-ray, optical properties and theoretical investigation', J. Mol. Struct., 1195: 32-42. https://doi.org/10.1016/j.molstruc.2019.05.109
- Udayakumar, Mani, Kothandapani Jagatheeswaran, Subramaniapillai Selva Ganesan, Natarajan S. Venkataramanan, Shankar Madan Kumar, Kullaiah Byrappa, and Subbiah Thamotharan. 2017. 'Investigation of 9-(2-hydroxy-4,4-dimethyl-6-oxocyclohex-1-en-1-yl)-3,3-dimethyl-2,3,4,9-tetrahydro-1H-xanthen-1-one: Crystal structure, AIM and NBO analysis', J. Mol. Struct., 1133: 510-18. https://doi.org/10.1016/j.molstruc.2016.11.082
- Venkatesan, P., M. Cerón, P. Ceballos, E. Pérez-Gutiérrez, S. Thamotharan, and M. J. Percino. 2019. 'Experimental study and DFT calculation for the strength of intermolecular interactions in Schiff base with the phenylarsonic acid scaffold', J. Mol. Struct., 1196: 306-22. https://doi.org/10.1016/j.molstruc.2019.06.073
- Venkatesan, P., M. Cerón, S. Thamotharan, F. Robles, and M. J. Percino. 2018. 'Quantitative analysis of weak non-covalent interactions in (Z)-3-(4-halophenyl)-2-(pyridin-2/3/4-yl)acrylonitriles', CrystEngComm, 20: 2681-97. DOI https://doi.org/10.1039/C7CE02096A
- Venkatesan, P., V. Rajakannan, N. S. Venkataramanan, A. Ilangovan, T. Sundius, and S. Thamotharan. 2016. 'Structural investigation of (2E)-2-(ethoxycarbonyl)-3-[(4-methoxyphenyl)amino]prop-2-enoic acid: X-ray crystal structure, spectroscopy and DFT', J. Mol. Struct., 1119: 259-68. https://doi.org/10.1016/j.molstruc.2016.04.090
- Venkatesan, P., S. Thamotharan, A. Ilangovan, H. Liang, and T. Sundius. 2016. 'Crystal structure, Hirshfeld surfaces and DFT computation of NLO active (2E)-2-(ethoxycarbonyl)-3-[(1-methoxy-1-oxo-3-phenylpropan-2-yl)amino] prop-2-enoic acid', Spectrochim. Acta - A: Mol. Biomol., 153: 625-36. https://doi.org/10.1016/j.saa.2015.09.002.
- Venkatesan, Perumal, Subbiah Thamotharan, Rajendran Ganesh Kumar, and Andivelu Ilangovan. 2015. 'Invariant and variable intermolecular interactions in functionalized malonic acid half-esters: X-ray, Hirshfeld surface and PIXEL energy analyses', CrystEngComm, 17: 904-15. https://doi.org/10.1039/C4CE02125H
- Venkatesan, Perumal, Subbiah Thamotharan, M. Judith Percino, and Andivelu Ilangovan. 2021a. 'Crystal Packing Modulation of the Strength of Resonance-Assisted Hydrogen Bonds and the Role of Resonance-Assisted Pseudoring Stacking in Geminal Amido Esters: Study Based on Crystallography and Theoretical Calculations', Cryst. Growth Des., 21: 779-98. https://doi.org/10.1021/acs.cgd.0c01010
- Venkatesan, Perumal, Subbiah Thamotharan, M. Judith Percino, and Andivelu Ilangovan. 2021b. 'Intramolecular resonance assisted N–H⋅⋅⋅O=C hydrogen bond and weak noncovalent interactions in two asymmetrically substituted geminal amido-esters: Crystal structures and quantum chemical exploration', J. Mol. Struct., 1246: 131210. https://doi.org/10.1016/j.molstruc.2021.131210
- Wood, Peter A., Joshua J. McKinnon, Simon Parsons, Elna Pidcock, and Mark A. Spackman. 2008. 'Analysis of the compression of molecular crystal structures using Hirshfeld surfaces', CrystEngComm, 10: 368-76. https://doi.org/10.1039/B715494A
Scheme 1 is available in the Supplementary Files section
No competing interests reported.
- Scheme1.png
Scheme 1. Synthesis of 2-bromo-4,6-bis(dibromoacetyl)resorcinol (I)
- Supplementarydata.docx
{kind=link}