Background: Frontotemporal dementia type 3 (FTD3) caused by a point mutation in the charged multivesicular body protein 2B (CHMP2B), affects mitochondrial ultrastructure and function as well as endosomal-lysosomal fusion in neurons. However, there is a critical knowledge gap in understanding how mutations in CHMP2B affect astrocytes. Hence, we investigated the disease mechanisms in astrocytes derived from hiPSC with mutations in CHMP2B and their impact on neurons.
Methods: To dissect the astrocyte-specific impact of mutant CHMP2B expression, we generated astrocytes from human induced pluripotent stem cells (hiPSCs) from FTD3 patients and their CRISPR/Cas 9 gene edited isogenic controls and produced heterozygous and homozygous CHMP2B-mutant hiPSC via CRISPR/Cas 9 knock-in gene editing. Additionally, we confirmed our findings in CHMP2B mutant mice. The hiPSC were subjected to astrocyte differentiation and the mutation dependent effects were investigated using immunocytochemistry, western blot, cytokine assays, transmission electron microscopy, RNA-sequencing and gas chromatography-mass spectrometry. Finally, neurons were exposed to conditioned media of mutant astrocytes and viability, growth and motility were measured.
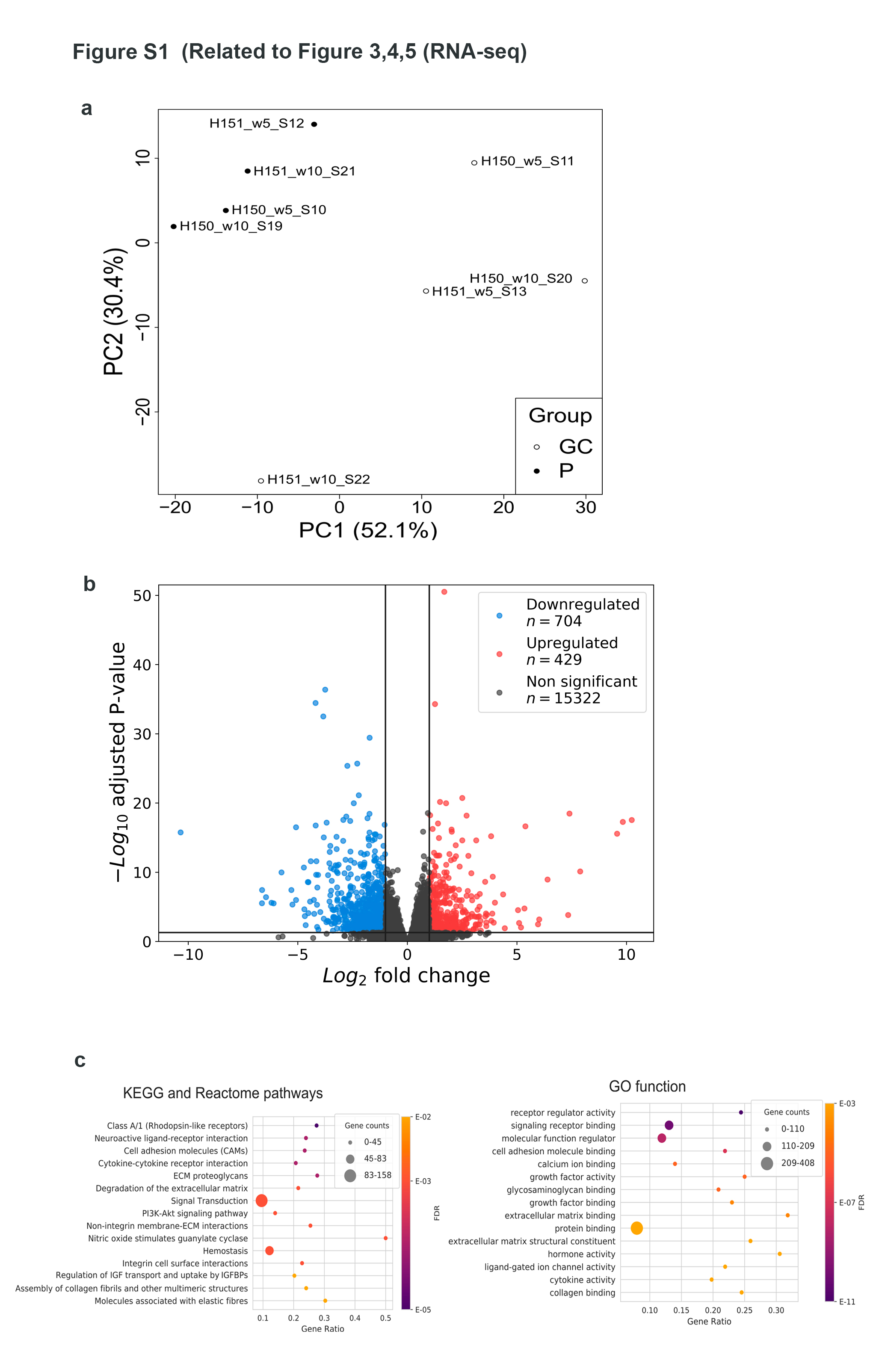
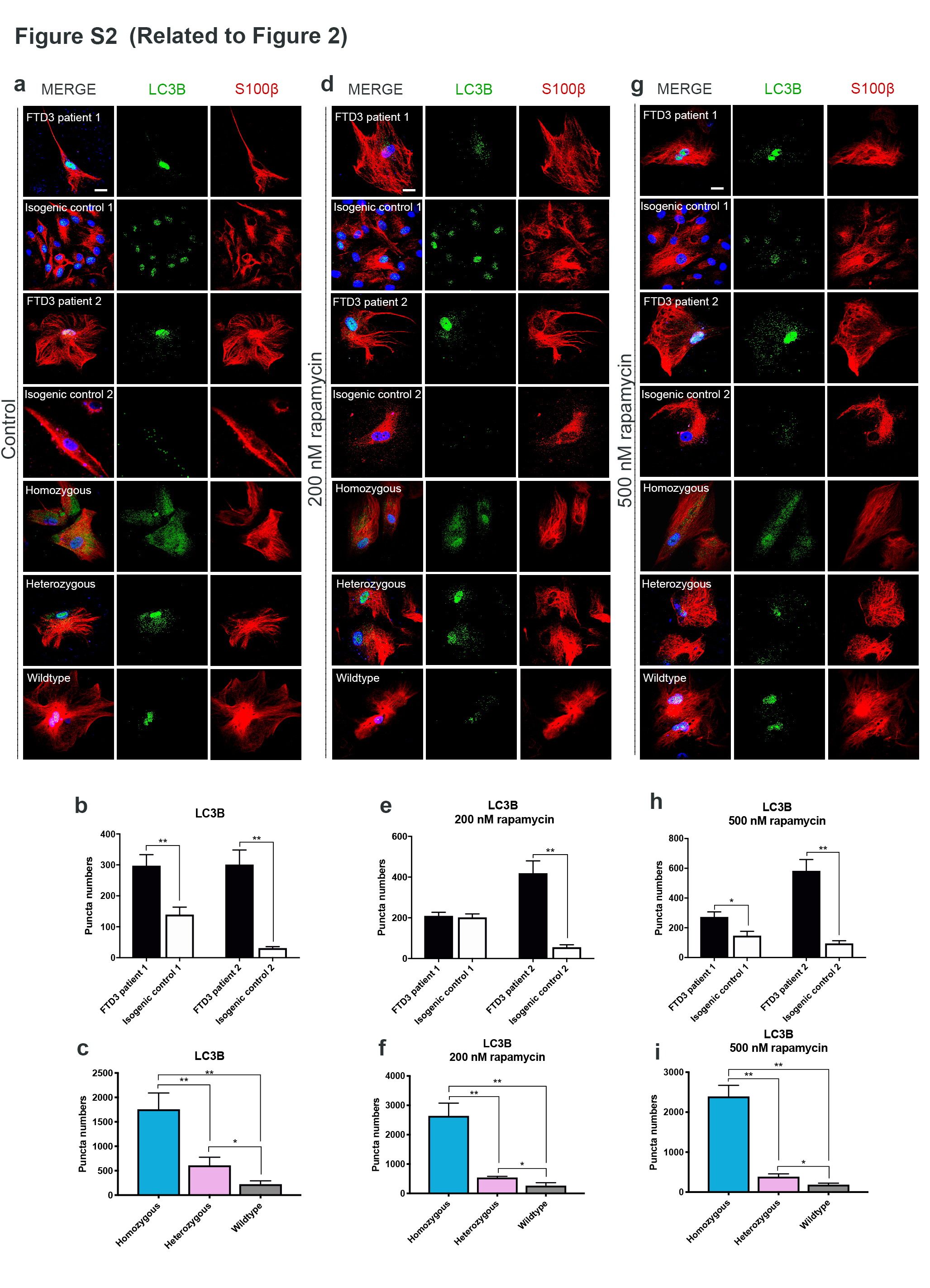
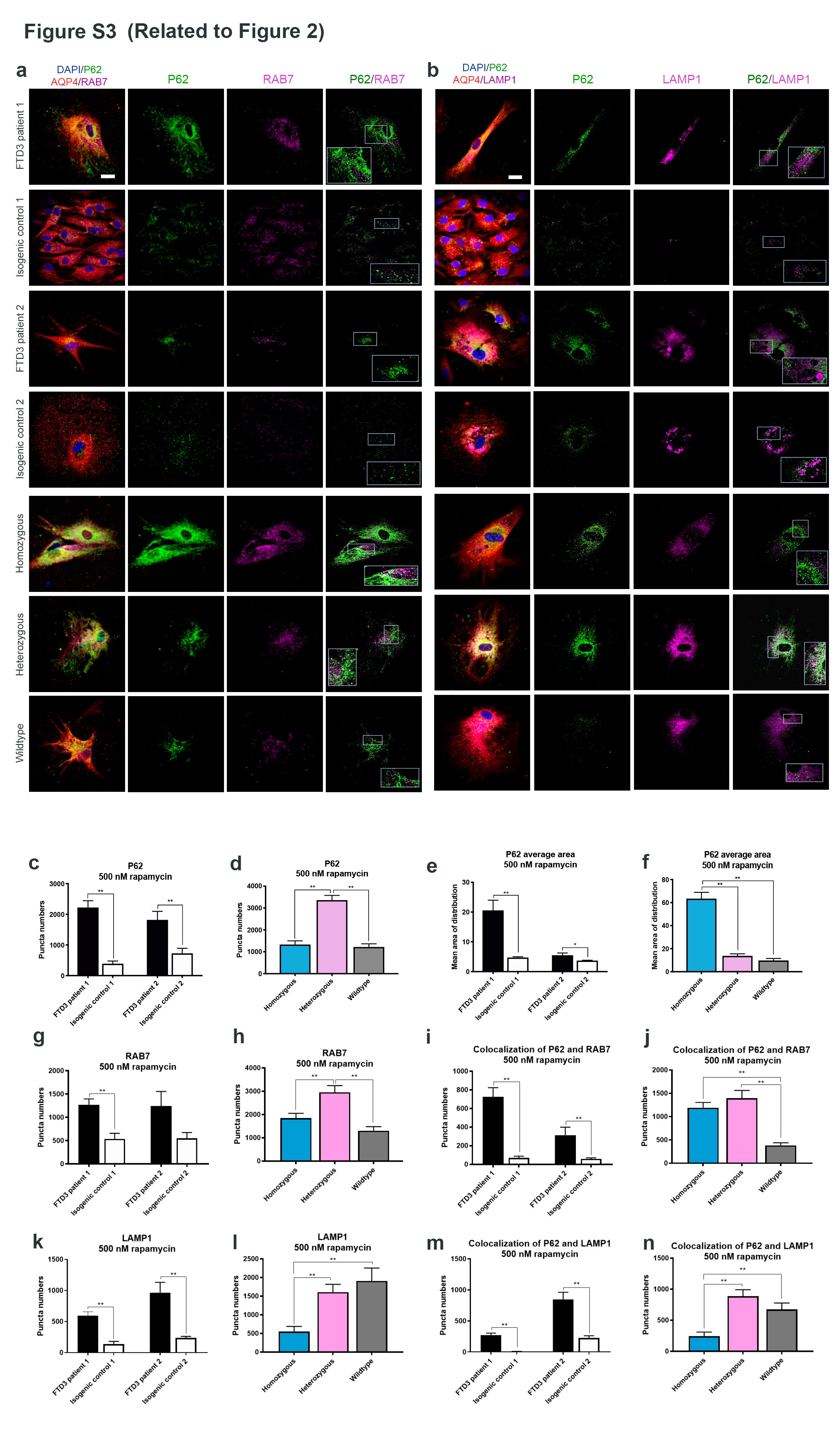
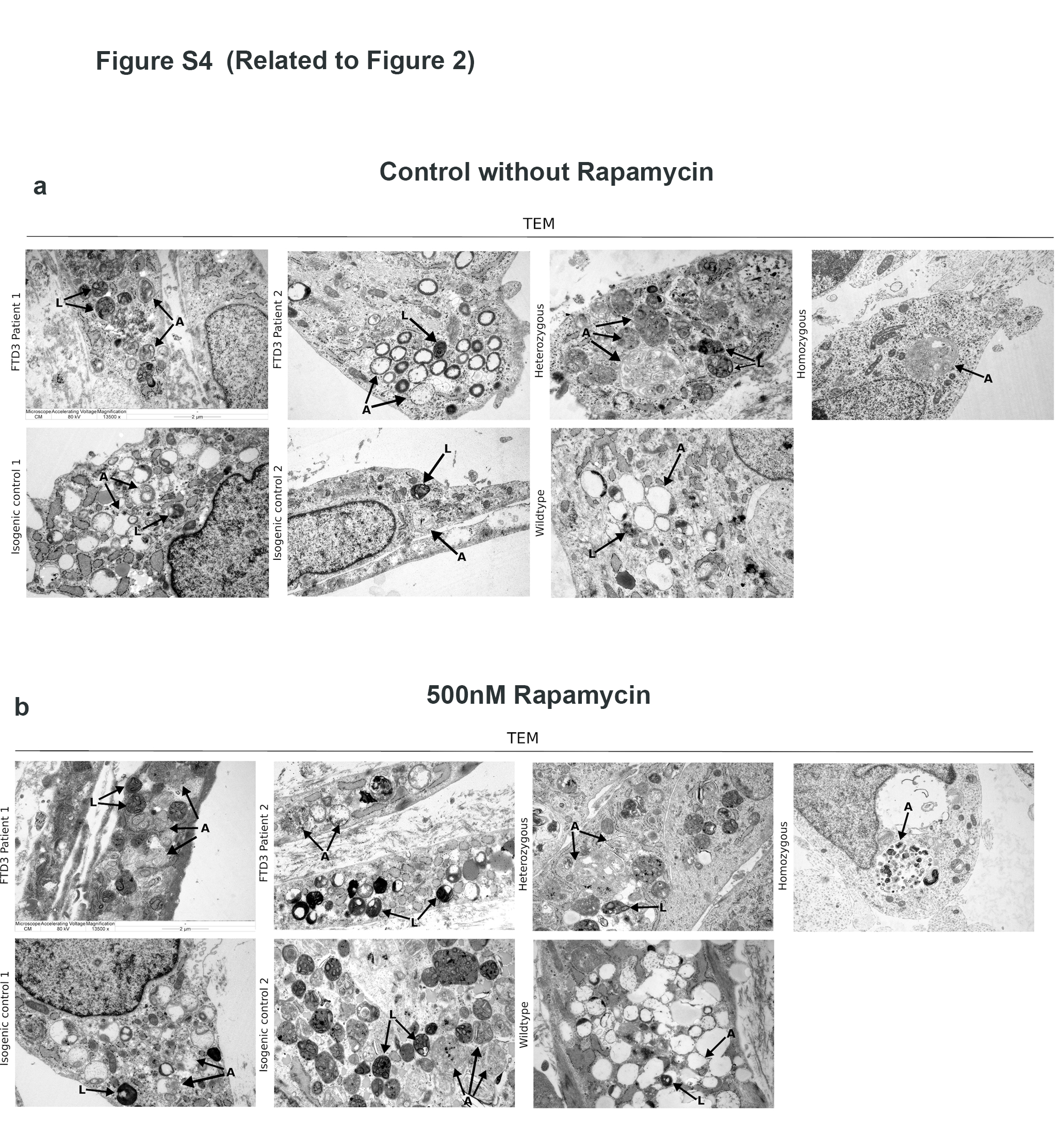
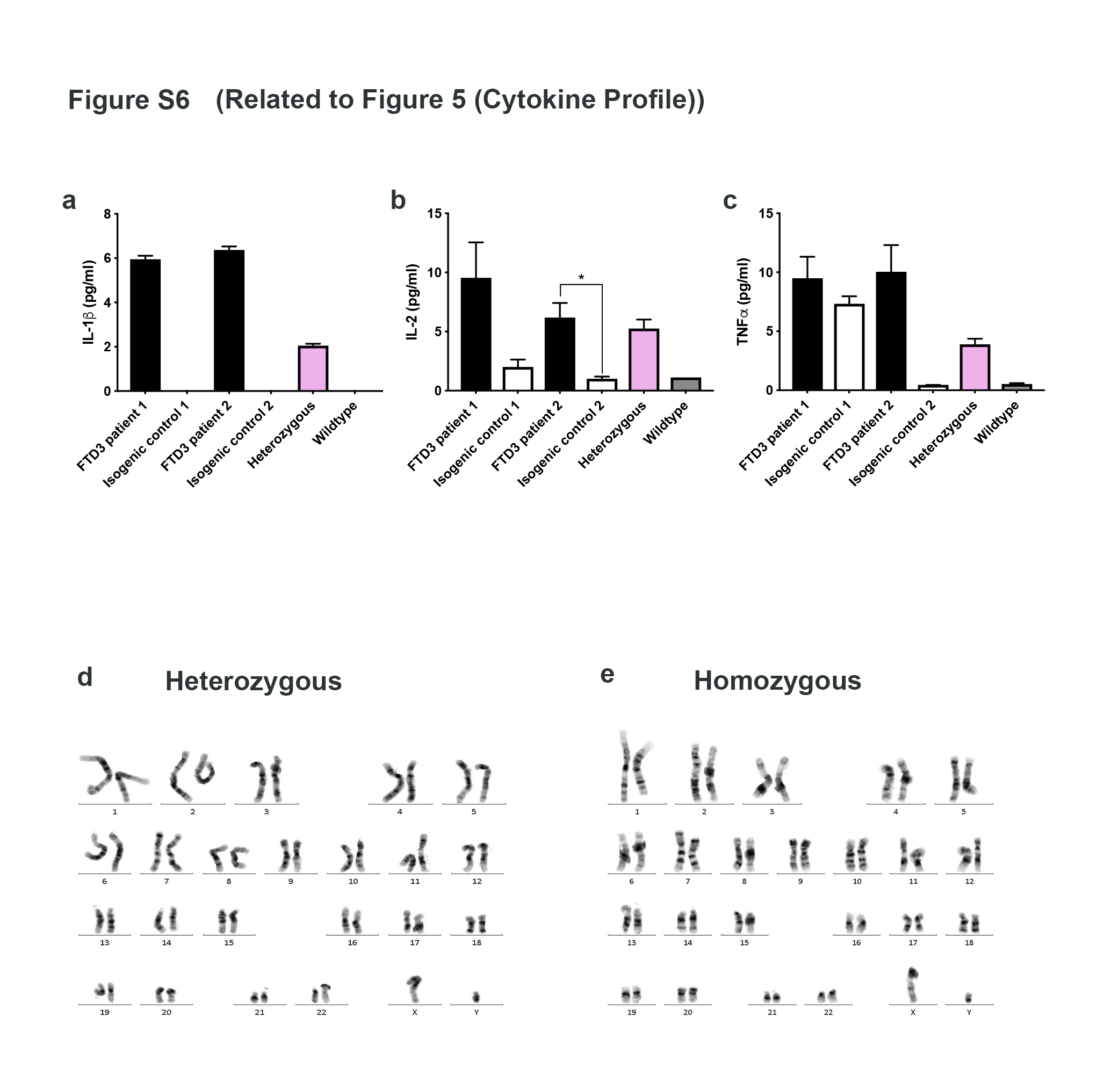
Results: To dissect the astrocyte-specific impact of mutant CHMP2B expression, we generated astrocytes from human induced pluripotent stem cells (hiPSCs) and confirmed our findings in CHMP2B mutant mice. Our findings include perturbed mitochondrial dynamics with impaired glycolysis, increased reactive oxygen species and elongated mitochondrial morphology, indicating increased mitochondrial fusion in FTD3 astrocytes. Furthermore, we identified a shift in astrocyte homeostasis triggering a reactive astrocyte phenotype and increased release of toxic cytokines. This cumulates in NF-kB pathway activation with increased production of CHF, LCN2 and C3, which cause neurodegeneration. The neurotoxic effect was investigated by exposing hiPSC-derived neurons to astrocyte-conditioned media, which severely reduced neurite outgrowth capacities. Rescue experiments targeting ROS could restore ROS levels back to normal levels, indicating that the impaired removal of abnormal mitochondria triggers the pathological cascade in CHMP2B mutant astrocytes culminating in the formation of neurotoxic reactive astrocytes.
Conclusion :Our data provide mechanistic insights into how defective mitophagy causes impaired mitochondrial fission, leading to the adoption of reactive astrocyte properties with increased cytokine release, NFkB activation and elevated expression of neurotoxic proteins in FTD3.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}