Bioinformatics Analysis
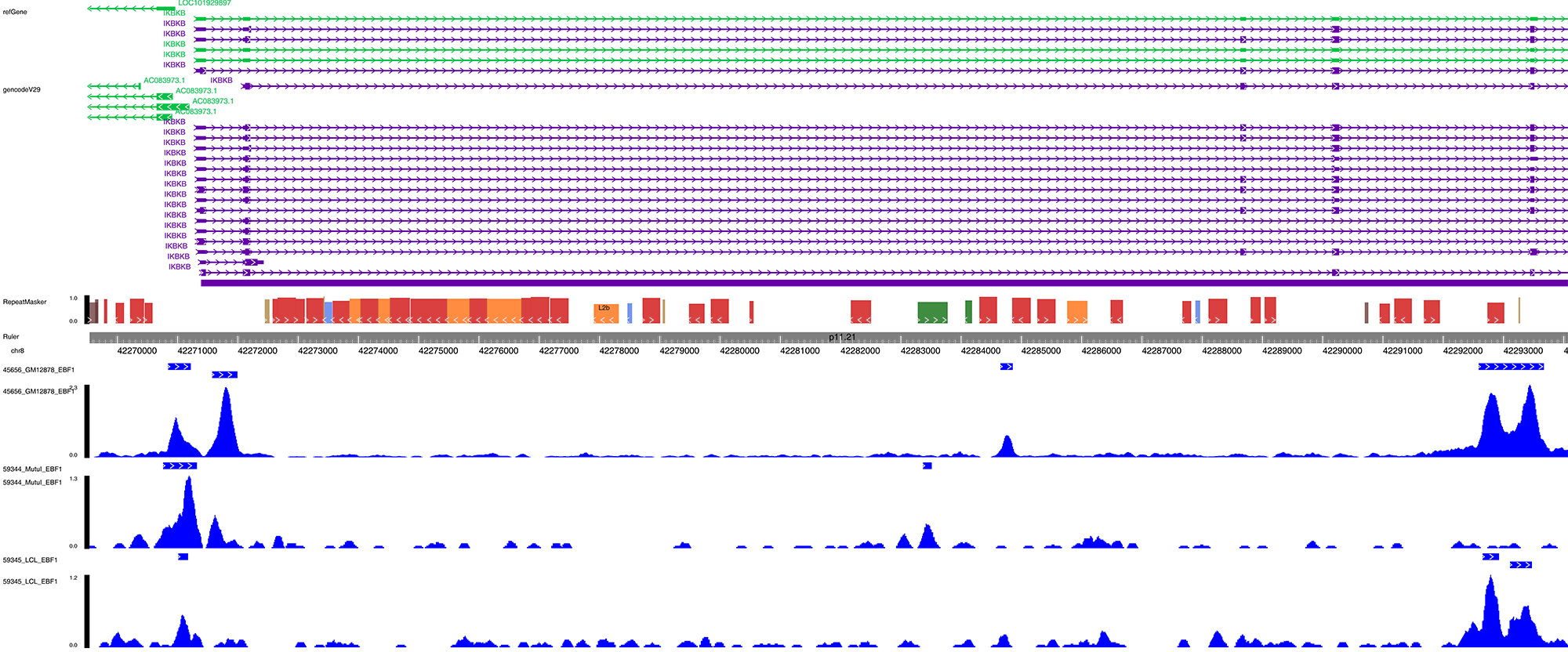
The Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) was searched for OA datasets. Microarray datawas collected from GEO datasets (GSE70362, GSE67566 and GSE116726). Raw files of microarray data were downloaded and normalised using a robust multiarray averaging method with ‘affy’ and ‘simpleaffy’ packages of ‘R’ software (www.R-project.org/). The processed gene expression matrix is provided in Supplementary Table S1, Supplementary Table S2, and Supplementary Table S3. The ‘pheatmap’ and ‘ggplot2’ packages of ‘R’ were applied to visualize the data. The enrichment analyses was calculated by datasets of GO (geneontology.org). Cytoscape (cytoscape.org/) was used to map the regulatory network based on the base pairing predicted by Starbase (starbase.sysu.edu.cn/) (Supplementary Table S4). The data of ChIP-seq was collected from Cistrome Data Browser (cistrome.org/db/#/). After searching for the keyword ‘EBF1’, data was analyzed in the current study (Fig. S1).
Clinical samples
Between October 2019 and March 2022, we procured NP tissues from a cohort of patients who had undergone Transforaminal Lumbar Interbody Fusion (n = 33, mean age 66.8 ± 2.1 years). Additionally, we obtained NP tissues from a group of patients who had undergone percutaneous endoscopic lumbar discectomy without IDD (n = 6, mean age 32.7 ± 2.5 years). Following surgical procedures, tissue samples were cryopreserved in liquid nitrogen prior to experimentation. A subset of tissues from healthy patients were subjected to direct digestion for subsequent cell culture.
Real-time PCR Analysis
The total RNA was isolated by RNeasy Mini Kit (74106, Qiagen, Valencia, CA) from NP tissues and cultured NPC. The cDNA was synthesized by QuantiTect Reverse Transcription Kit (205314, Qiagen, Valencia, CA). Quantitative real-time PCR (q-PCR) was done by using QuantiFast SYBR Green PCR Kit (204056, Qiagen, Valencia, CA). Relative RNA expression was calculated using the 2−ΔΔCTmethod with normalization to U6 small nuclear RNA. Each experiment was performed in triplicate.
NPC culture
We extracted NPC from normal patients’ NP tissues as previously described. Briefly, NP tissue was cut into as small pieces as possible and then digested with 0.1% trypsin (15400054, Gibco Company, USA) for 20 minutes. Thereafter, it was digested with a Type II collagenase (17101015, Gibco Company, USA) in Dulbecco’s modified Eagle’s medium (DMEM, 11965092, Gibco Company, USA) in the water bath at 37°C for 30 minutes. Undigested tissue was filtered using a 40 nm strainer. The NPC were then cultured in DMEM containing 10% fetal bovine serum (30044333, Gibco Company, USA). The second and third passage cultured NPC were used for the experiment.
Cell transfection
The cDNA of EBF1, circEYA3 and IKKβ were cloned into the multiple cloning site of the pcDNA3.1 vector (Invitrogen, USA). MiR-196a-5p mimic, negative control oligonucleotides (miR-NC), miR-196a-5p inhibitor, negative control oligonucleotide (NC inhibitor), small interfering RNA of EBF1, circEYA3 or IKKβ (siEBF1, sicirc-1, sicirc-2, sicirc-3, siIKKβ), scramble siRNA of EBF1 circEYA3 or IKKβ (siSCR) were purchased from RiboBio (Guangzhou, China). Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) was used for the transfection assay in 6-well plate with seeded chondrocytes. Q-PCR was used to detect the expression level of mRNA. Transfection efficiency was detected by fluorescence microscope (OLYPAS, Japan). After 48h, chondrocytes were stimulated with IL-1β (10ng/ml) for the 24h and used for further analysis.
Western blot assay
Cytoplasmic protein and nuclear protein were exacted using a Nuclear and Cytoplasmic Extraction Reagents kit (78835, Thermo Fisher Scientific, USA). The protein concentration was measured by the BCA Assay Kit (23223, Thermo Scientific, USA). The protein was electrophorsed with 10% SDS-PAGE gels, and subsequently transferred to the polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA, USA). Specific primary antibodies (ab181602, ab32535, ab39012, ab188570, ab214429, ab32561, ab76429, ab124957, ab32536, ab32511, Abcam, Cambridge, UK) (PA5-61136, Invitrogen, USA) were co-incubated with the membrane overnight at 4°C. Rewarming the membranes for 2h, and then incubated with anti-rabbit IgG (ab97051, Abcam, Cambridge, UK) at 37°C for 2h. ECL Western blot kit (32209, Thermo Fisher Scientific, USA) were used to detect the bands. GAPDH and Lamin B were used as control.
Immunofluorescence (IF) staining
The NPC were cultured in a 12-well plate and stimulated with IL-1β for 24h. Primitively, cells were fixed with 4% paraformaldehyde for 20 min. Subsequently, the cells were treated with 0.2% Triton X-100 for 3 min, and blocking with 5% BSA for 1h. The cells were incubated with primary antibody (Abcam, Cambridge, UK) overnight at 4℃. The chondrocytes were washed with PBS and incubated with goat anti-rabbit IgG (SA00013-4, SA00013-2, Proteintech, China) for 1h at room temperature. Lastly, DAPI(4',6- diamidino-2-phenylindole) (c0065, Solarbio, China) was used for nuclear staining. The fluorescence was observed by with fluorescence microscope (OLYPAS, Japan).
Flow cytometry
PE Annexin V Apoptosis Detection Kit I (559763, BD Pharmingen, USA) was used to determine the apoptotic rate of treated chondrocytes. Subsequently, the results were analyzed with the fluorescence-activated cell sorting (FACS) flow cytometer (BD Biosciences, USA). Each experiment was performed in triplicate.
Dual luciferase reporter gene assay
The pmirGLO Dual-Luciferase miRNA Target Expression Vector was obtained from GenePharma (China). The amplified DNA sequences were cloned to the pmirGLO reporter plasmid to form wild type EBF1 3′-UTR (WT) and mutated EBF1 3′-UTR (MUT) luciferase vectors. The pmiRGLO luciferase reporter vectors of the circEYA3 were constructed as above. Human chondrocytes were plated (5× 104 cells per well) in 24-well plates overnight. Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) was used to transfect the NPC with plasmid and miR-196a-5p mimic or the control. The luciferase activity was measured by the dual-luciferase reporter gene assay system (E1910, Promega, USA) after 48h. Data were shown as the mean value ± SD and each experiment was performed in triplicate.
RNA immunoprecipitation (RIP) assay
Magna RIPTM RNA Binding Protein Immunoprecipitation Kit (17–700, Millipore, USA) was used to conduct the RIP assay. The endogenous miR-196a-5p, combined with circEYA3, was pulled down. Briefly, cultured chondrocytes were collected and resuspended in RIPA buffer (R0020, Solarbio, China). Then incubated the cell extracts with RIP buffer containing magnetic beads conjugated with human anti-Ago2 antibody (07-590, Millipore, USA) or mouse IgG (CBL610, Millipore, USA) negative control overnight at 4°C. The next morning, proteinase K was used to co-incubate with the magnetic beads after washing three times. Subsequently, total RNAs was isolated from the extracts using the TRIzol reagent. The relative enrichment of circEYA3 and miR-196a-5p were determined by RT-qPCR analysis.
Fluorescence in situ hybridization (FISH) assays
The FISH assays were done by FISH kit (F03402, GenePharma, China). In Brief, the probes specific to circEYA3 and miR-196a-5p were added to NPC and then pre-denatured at 78℃ for 5 minutes. Then, hybridization was carried out at 42℃ overnight. DAPI (c0065, Solarbio, China) was used to counterstaining the nuclei with 20 min in darkness. The sample was scanned and photographed under Olympus microscope.
Chromatin Immunoprecipitation (ChIP)
Chip assay was performed using the EZ-Magna ChIP kit (17-10086, EMD Millipore, GER). Human chondrocytes were fixed with 4% paraformaldehyde and incubated with glycine for 10 min to generate DNA–protein cross-links. Then, the cells were lysed with Cell Lysis Buffer and Nuclear Lysis Buffer and sonicated to generate chromatin fragments of 400–800 bp. The lysates were immunoprecipitated with Magnetic Protein A Beads conjugated with EBF1 antibody (ab108369, Abcam, Cambridge, UK) or IgG. Finally, the precipitated DNA was analyzed by PCR.
Rat model of IDD
All animal experimental procedures were ratified by the Animal Ethics Committee of Dalian Medical University. The study was experimented in strict compliance with the Guidelines for the Care and Use of Laboratory Animals published in 2011. The rats were purchased from the Experimental Animal Center of Dalian Medical University. The IDD model was made as described previously. Briefly, the rats were placed in the supine position to locate the lumbar disc behind the bifurcation of the inferior vena cava after anesthesia. The annulus fibrosus is then punctured with a syringe and left for 1 minute. Finally, the rat abdomen was sutured layer by layer. MiR-NC, miR-196a-5p agomir (50µM) and lentivirus (1×109 PFU, 20µl) expressing EBF1 or circEYA3 were injected into the lumbar disc of recipient rats (20µL per joint per rat twice a week for 4 weeks) (n = 12 per group). Eight weeks after surgery, rats lumbar were scanned and used for subsequent experiments.
Histology staining
The rats’ lumbar were fixed in 4% paraformaldehyde for paraffin. Decalcification of tissue using EDTA decalcification solution for two months. The section was pre-treated with drying, deparaffining and rehydrating. Then cut the tissues into 4µm slices. Safranin-O & fast green-stained (G1371, Solarbio, China) sections were used to evaluate the degeneration of the NP and the Morphology of the lumbar.
Statistical analysis
SPSS 17.0 software was used to analyze the data. Data were presented as means ± standard deviation (SD). Student ’s t-test was used to compare the significant difference of two groups. Standard deviation (SD) represented the variation of data values. The one-way analysis of variance (ANOVA) was used to determine the significant difference of multiple groups. Pearson Correlation Coefficient was used to identify the correlation. Each experiment was carried out thrice at least. Statistical significance was defined as P value < 0.05.
{kind=link}