STK11 loss alters tumor-intrinsic cytokine expression
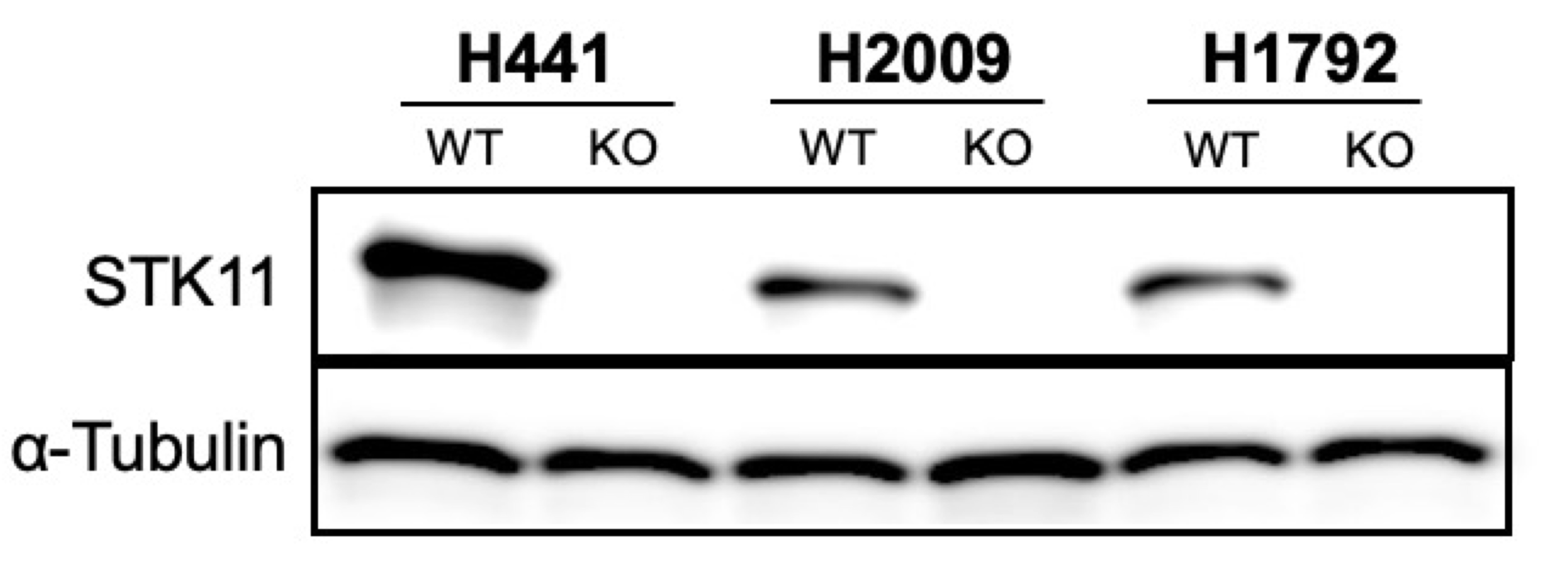
We knocked-out STK11 in three genetically independent human KRAS-driven LUAD cell lines that normally harbor intact STK11 alleles: NCI-H2009, NCI-H441 and NCI-H1792. STK11 loss was validated by Western Blot analysis (Fig. 1A and Supplemental data). Based on studies reporting a correlation between Stk11 loss and Il-6 upregulation in mouse models of Kras-driven lung cancers in vivo12, we compared IL-6 expression between STK11 WT (aka “Parent”) and matched STK11-KO human LUAD cells using qRT-PCR. Unexpectedly, under standard culture conditions no difference in IL-6 expression was detected between the Parent and STK11-KO cells (Fig. 1B, “+Glutamine”). However, significant STK11-loss-dependent IL-6 upregulation was observed when cells were cultured under conditions of nutrient stress, achieved via glutamine depletion (Fig. 1B, “-Glutamine”). The rationale for evaluating nutrient stress as a variable was based on evidence that STK11 functions as a nutrient sensor to regulate metabolic homeostasis19–21. We reasoned STK11 loss might be irrelevant when cells are grown in standard media as nutrients are in excess. Given that tumor microenvironments in vivo are characterized by nutrient stress22–24, we used glutamine depletion to simulate nutrient-deprivation in vitro.
Next, to comprehensively characterize STK11-loss-dependent transcriptional changes, we expanded our analyses and performed whole transcriptome sequencing comparing standard media to glutamine depletion. In standard media, relatively few genes differed between parent and STK11-KO cells (Fig. 1C, +Glutamine; H2009: 1100 DEGs, H441: 928 DEGs). In contrast, when comparing both H2009 and H441 parent lines with their paired STK11-KO lines following glutamine depletion we identified 7453 and 5202 differentially expressed genes (DEGs) respectively (Fig. 1C; -Glutamine). This marked STK11-loss-dependent transcriptional impact indicates STK11 plays a critical and generalizable role in regulating transcription in response to nutrient stress. We then performed Gene Set Enrichment Analysis (GSEA)25 on the DEGs for both H2009 and H441 cell lines and found significant associations between STK11 loss and altered tumor-intrinsic cytokine signaling, specifically upregulation of genes within the Gene Ontology (GO) term “Cytokine Activity” (GO: 0005125) (Fig. 1D). Of the upregulated genes in this curated list, 9 were shared between the H2009 and H441 cell lines, suggesting overlapping regulatory pathways. Intriguingly, these overlapping genes consist of effectors previously associated with cancer progression, immune evasion, and therapy resistance26–28. For example, both IL-6 and CXCL8 are reported to be elevated in KRAS-driven STK11-null LUADs and proposed to promote tumor immune evasion29–31. Similarly, CXCL2 is known to drive neutrophil recruitment, a phenotype associated with “cold” tumor immune microenvironments27. Finally, BMP2 expression is correlated with metastatic burden and STK11 loss in lung cancer and mediates activation of SMAD transcription factors32,33, which are known YAP1 binding partners34.
YAP1 transcriptional activation occurs in LUAD cell lines lacking STK11
In addition to GSEA, we also performed pathway enrichment using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database35. This approach revealed several significantly enriched networks in STK11-KO cells relative to matched parental lines (Fig. 1E-F). Consistent with prior published reports, both focal adhesion and HIF-1 pathways were over-represented in cells lacking STK1136,37. In addition, NF-kappa B signaling, TNF signaling, chemokine signaling and HIPPO signaling were significantly enriched in STK11-KO cells. We chose to focus on the HIPPO pathway as STK11 has previously been implicated in HIPPO regulation via direct activation of MARK family kinases and subsequent modulation of YAP1 activity13. YAP1-mediated transcriptional activation is controlled in part via cytosolic sequestration; a kinase-dependent process regulated by activation of the HIPPO cascade16. Utilizing a curated list of YAP1 transcriptional target genes13, we repeated GSEA and found a significant positive correlation between STK11 loss and enhanced expression of YAP1 target genes in both the H2009 and H441 cell lines (Fig. 1G).
STK11 loss correlates with increased YAP1 protein levels
STK11 has previously been proposed to indirectly modulate the HIPPO/YAP1 axis via MARK activation, ultimately promoting YAP1 sequestration and degradation13. We therefore hypothesized that STK11 loss would result in increased YAP1 protein due to enhanced protein stabilization (Fig. 2A). Western blot analysis comparing whole cell extracts from parent and STK11-KO LUAD cell lines support this assertion, showing a ~ 2-fold increase in relative YAP1 abundance (Fig. 2B), an observation supported by prior studies in mice13. Interestingly, this difference occurs only at the protein level, as YAP1 transcript levels remain unchanged, supporting our hypothesis that STK11 loss results in YAP1 protein stabilization (Fig. 2C). Nuclear and cytosolic fractionation analyses further demonstrate that increased YAP1 protein levels are not isolated to either compartment but increased throughout cells lacking STK11. Upon glutamine deprivation, we observed increased YAP1 nuclear translocation in both parent and STK11-null cells, though the increase was more pronounced in the STK11-null cells (Fig. 2D-E). This data supports an STK11-dependent impact on global YAP1 protein abundance, including nuclear localization, which we posit drives changes in YAP1-mediated gene expression (Fig. 2A and Fig. 1G).
YAP1 antagonism partially restores cytokine expression profiles in STK11 deficient cells
To validate our pathway analyses we reasoned we could inhibit STK11-loss-dependent cytokine induction following glutamine depletion by blocking the downstream signaling networks responsible. To examine the role of YAP1 in driving this phenotype, we engineered STK11/YAP1 double knockouts in both H2009 and H441 LUAD cell lines (Fig. 3A). Our data demonstrate significantly less IL-6, CXCL8 and CXCL2 expression in the STK11/YAP1 double KO lines compared with STK11-KO lines following glutamine depletion (Fig. 3B). Importantly, these changes were mirrored by levels of secreted IL-6 and CXCL8 protein levels measured by ELISA (Fig. 3C). YAP1-KO alone had no impact on expression of these cytokines, regardless of glutamine availability, demonstrating the necessity of STK11 loss in producing this phenotype (Fig. 3B).
After establishing YAP1 functions downstream of STK11 and is at least in part responsible for the increased cytokine expression occurring in STK11-KO cells following glutamine depletion, we next sought to phenocopy YAP1 KO via pharmacologic antagonism of YAP1 with verteporfin (VP)38. One mechanism by which VP is known to alter YAP1 activity occurs via physically disrupting the interaction between YAP1 and members of the TEAD transcription factor family38. Our data clearly show that the STK11-loss-dependent upregulation of IL-6 and CXCL8 upon glutamine depletion is blunted by VP treatment (Fig. 3D). Interestingly, this affect does not extend to CXCL2 (Fig. 3D). Together these results support CXCL8 and IL-6 expression are likely regulated, at least in part, by YAP1/TEAD interactions. The fact that CXCL2 expression is reduced upon YAP1 genetic ablation, but not VP treatment, was unexpected and suggests YAP1’s impact on CXCL2 expression may be independent of TEAD. YAP1 is known to interact with many transcription factors, including SMAD family members and the b-catenin/TBX5 complex34. We think it likely that YAP1’s impact on CXCL2 expression relies on a transcription factor other than a TEAD family member, which is why genetic ablation of YAP1 results in altered expression, whereas TEAD dissociation with VP does not. Whether this definitively explains the discrepancy in our CXCL2 data awaits further investigation but remains a favored hypothesis.
YAP1 ablation restores gene expression profiles in STK11 deficient cells
To define the transcriptome-wide impact of YAP1 KO in STK11 deficient cells, we performed RNA-seq on H2009 cells following 24hrs in either standard or glutamine depleted media. In standard media, few genes differed between STK11-KO and STK11/YAP1 double KO cells (Fig. 4A, +Glutamine; 733 DEGs). Compared with the H2009 parent line, similar numbers of DEGs were detected in the STK11/YAP1 double KO as were seen in the STK11 KO when grown in the absence of glutamine (Fig. 4A, -Glutamine; 7698 DEGs vs Fig. 1C, -Glutamine; 7453 DEGs). However, when the STK11/YAP1 double KO cells are compared directly with STK11 KO cells in the absence of glutamine, 4167 DEGs were detected (Fig. 4A, -Glutamine; 4167 DEGs). If YAP1 loss had no impact, we would predict no DEGs identified between these two conditions. The DEGs detected represent genes that still change upon glutamine depletion, but the magnitude of that change is significantly reduced in the absence of YAP1 indicating these genes are candidates for YAP1-mediated regulation. Kmeans clustering of genes differentially expressed between STK11-KO and STK11/YAP1 double KO cells reveled a large group of genes that, while still induced by glutamine depletion, were repressed relative to the induction observed in STK11-null/YAP1-competent cells (Fig. 4B; cluster 2, Red v Orange). GSEA performed on DEGs identified between H2009 STK11-KO and STK11/YAP1 double KO cells using the previously described curated YAP1 gene signature demonstrated a significant negative correlation, indicating gene repression in STK11/YAP1 double KO cells relative to STK11-KO/YAP1-intact cells (Fig. 4C). Specifically, 102 genes within the curated YAP1 signature exhibited reduced expression upon YAP1 ablation in STK11-KO cells, highlighted by dot plot analysis of the 17 genes identified in Fig. 1G, which show overlap in gene induction between H2009 and H441 cells upon STK11 ablation (Fig. 4C). We posit those genes demonstrating significant reduction in expression are regulated in part by YAP1. We also performed GSEA using the cytokine activity signature (GO: 0005125) previously described (Fig. 1D) and observed repression of 35 member genes upon YAP1 ablation in STK11-KO cells (Fig. 4D). Again, dot plot analysis highlights repression of a subset of these genes following YAP1 deletion in H2009 cells lacking STK11 (Fig. 4D). Taken together, these data support YAP1 antagonism as a strategy to curb expression of key genes, including immunomodulatory cytokines, in KRAS-driven STK11-null LUADs. We speculate a similar response in vivo would aid in transitioning immunologically “cold” tumor immune microenvironments to “hot”, potentiating the effectiveness of checkpoint inhibitor therapies such as anti-PD-1 monoclonal antibodies (Fig. 4E).
{kind=link}