The ALPL gene encoding TNSALP was located on chromosome 1p34–36. HPP is caused by loss of function mutations in the ALPL gene. The first case of HPP was reported in 1948 and the ALPL gene mutation was first reported in 1988 [1, 22]. HPP presents a heterogeneous phenotype ranging from life threatening to asymptomatic presentation. In general, the pattern of inheritance in HPP is autosomal recessive. However, HPP may also be transmitted by autosomal dominant and incomplete penetrance of dominant transmission [23]. The severe forms of HPP usually transmit by autosomal recessive, while milder forms of HPP more often present with autosomal dominant with variable expressivity. These heterogeneous clinical features and inheritance patterns result in the difficulty of timely diagnosis and challenging of genetic counselling [24, 25].
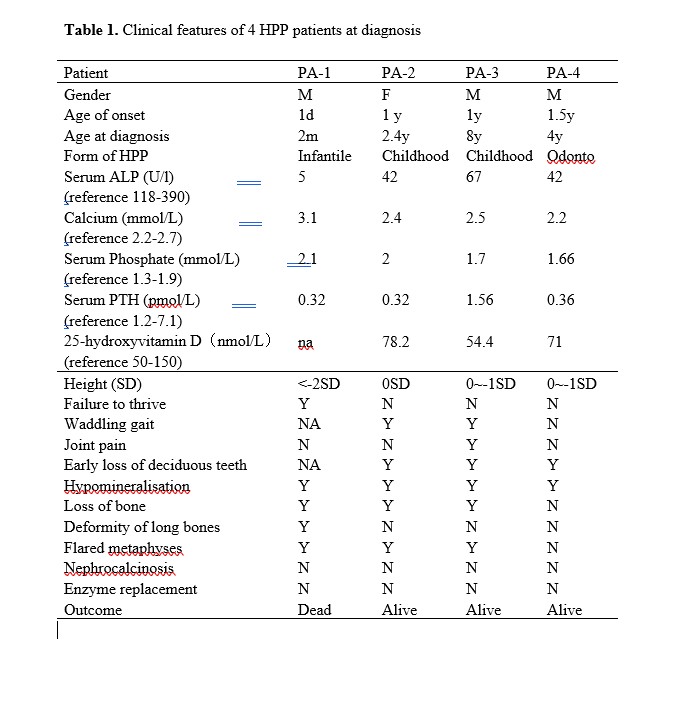
In the present study we investigated four Chinese children affected by three forms of HPP, which were lethal infantile, childhood and odonto HPP. All patients follow autosomal recessive inheritance pattern. They presented clinical symptoms early at 1 day old (patient 1) or at 1 year old (patient 2, 3 and 4). All four patients had low serum ALP activity. As reported, patients with perinatal or infantile HPP have high morbidity and mortality in the first 5 years of life [7]. Patient 1 was a typical infantile form of HPP who presented respiratory distress, nearly undetectable ALP activity and general skeletal hypomineralization. He died of respiratory failure at 3 months old, therefore was diagnosed as lethal infantile form of HPP. To the best we know, this was the first case of lethal infantile HPP reported in the Chinese population. Recent researches revealed that a rapidly worsening clinical course often occurs in prenatal [26] or infantile HPP mainly due to respiratory compromise [7]. However, further investigation is need to clarify the mechanism.
It has been reported that diagnostic delay is common due to limited awareness of HPP [27]. Patient 2 and 3 were Childhood HPP who presented premature loss of deciduous teeth and muscle weakness at 1 year old. Patient 3 also had joint swelling and bone pain starting at 1 year old, however, he was confirmed with HPP at 8 years old. Patient 4, although his early loss of deciduous teeth started at 1 year old, was diagnosed at 4 years old. These observation demonstrated that some HPP cases may not be recognized well and managed timely.
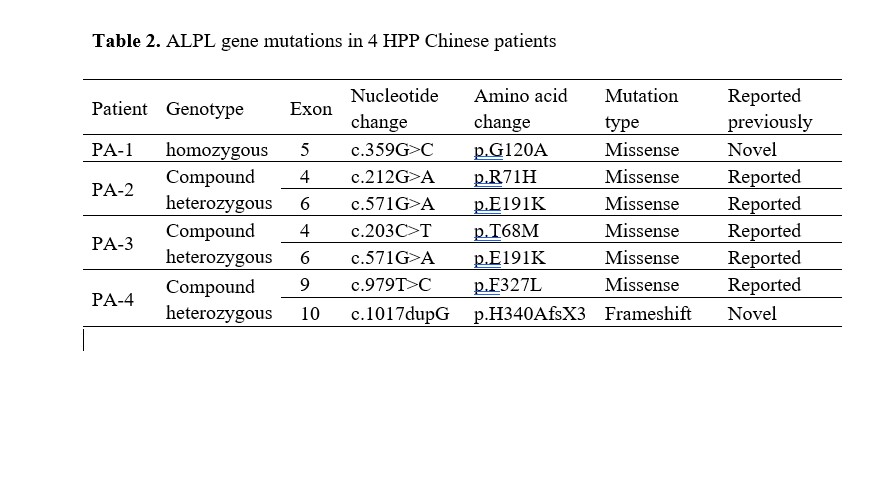
Molecular diagnosis provides great advantage to confirm the diagnosis of HPP. HPP is caused by a loss of function mutation in the ALPL gene encoding TNSALP. It has been reported that few mutations may be frequent in particular populations [24]. For instance, c.1559delT and p.F327L are two common mutations in the Japanese population, whereas c.571G > A (p.E191 K) is identified in half of European patients with moderate HPP [11, 28]. To elucidate the mutational characteristics of the ALPL gene in the Chinese population, we reviewed all reported Chinese HPP cases in the literature. We found that the most mutations in Chinese HPP patients were missense variants located in exon 5 in the ALPL gene. No frequent mutations were recognized (Figure 3). In the present study, six ALPL mutations, including five missense mutations and one splicing mutation, were identified. All of these mutations were first reported in the Chinese population.
It is mainly considered that the variety of ALPL mutations results in highly variable clinical expressivity, resulting in difficulty to assess the severity of a novel mutation in the ALPL gene [16]. The severity of HPP symptoms was correlated with the level of ALP activity affected by the ALPL mutation [29]. Severe HPP often exhibit very low level of ALP activity, whereas mild HPP usually retain most ALP activity. Severe form manifests early, whereas mild form may be diagnosed in adulthood [30]. Patient 1 was a homozygote of c.359G>C which has not been previously described in the literature. However, typical clinical features, very low level of serum ALP activity, overall hypomineralization and homozygote status all were consistent with severe infantile form of HPP, indicating the variant c.359G>C seems to be at a crucial position of ALP protein. Further investigation revealed that the variant c.359G>C (p.G120A) is located in the homodimer interface, a crucial site of secondary structure in the TNSALP and highly conserved throughout many species. Thus, the novel mutation c.359G>C was strongly indicated to be disease-causing and related to severe infantile form of HPP. Further functional study of the mutation c.359G>C is needed.
Patient 2, 3 and 4 were compound heterozygote in the ALPL gene. Patient 2, the childhood HPP, had been identified with two known pathogenic variants, c.212G>A (p.R71H) at exon 4 and c.571G>A (p.E191K) at exon 6[31, 32]. As another childhood HPP, Patient 3 carried the identical c.571G>A (p.E191K) variant of Patient 2 and another pathogenic c.203C>T (p.T68M) variant at exon 4 which was also reported previously [33]. Interestingly, the variant c.571G > A (p.E191 K) in Patient 2 and 3, was the common mutation reported in European population with moderate HPP. Patient 4 was the mildest odonto HPP in the present study. He demonstrated c.979T>C (p.G120A) and c.1017dupG (p. H340AfsX3). The c.979T>C (p.G120A) variant was pathogenic reported previously [34], whereas the novel variant c.1017dupG (p.H340AfsX3) was predicted to result in a frameshit and premature protein termination (p. H340AfsX3).
Some limitations exist in the present study. First, the number of HPP patients is not big enough to reveal the phenotype–genotype correlations. Second, PLP, the best markers of HPP, was not detected in the present study since the method is still not available in our center. Future study will be focused on collecting more data to reveal the correlation between phenotype and genotype of HPP in the Chinese population.
In the present study, we described the clinical and genetic characteristics of HPP in four unrelated Chinese pedigrees who were affected with infantile, childhood and odonto forms of HPP. All patients followed autosomal recessive inheritance pattern. Six mutations in the ALPL gene were identified including four known mutations and two novel mutations. The novel missense mutation (c.359G>C) caused the decrease of ALP activity and was related to lethal infantile form of HPP. The study expanded knowledge about the characteristics of HPP.
{kind=link}
{kind=link}
{kind=link}