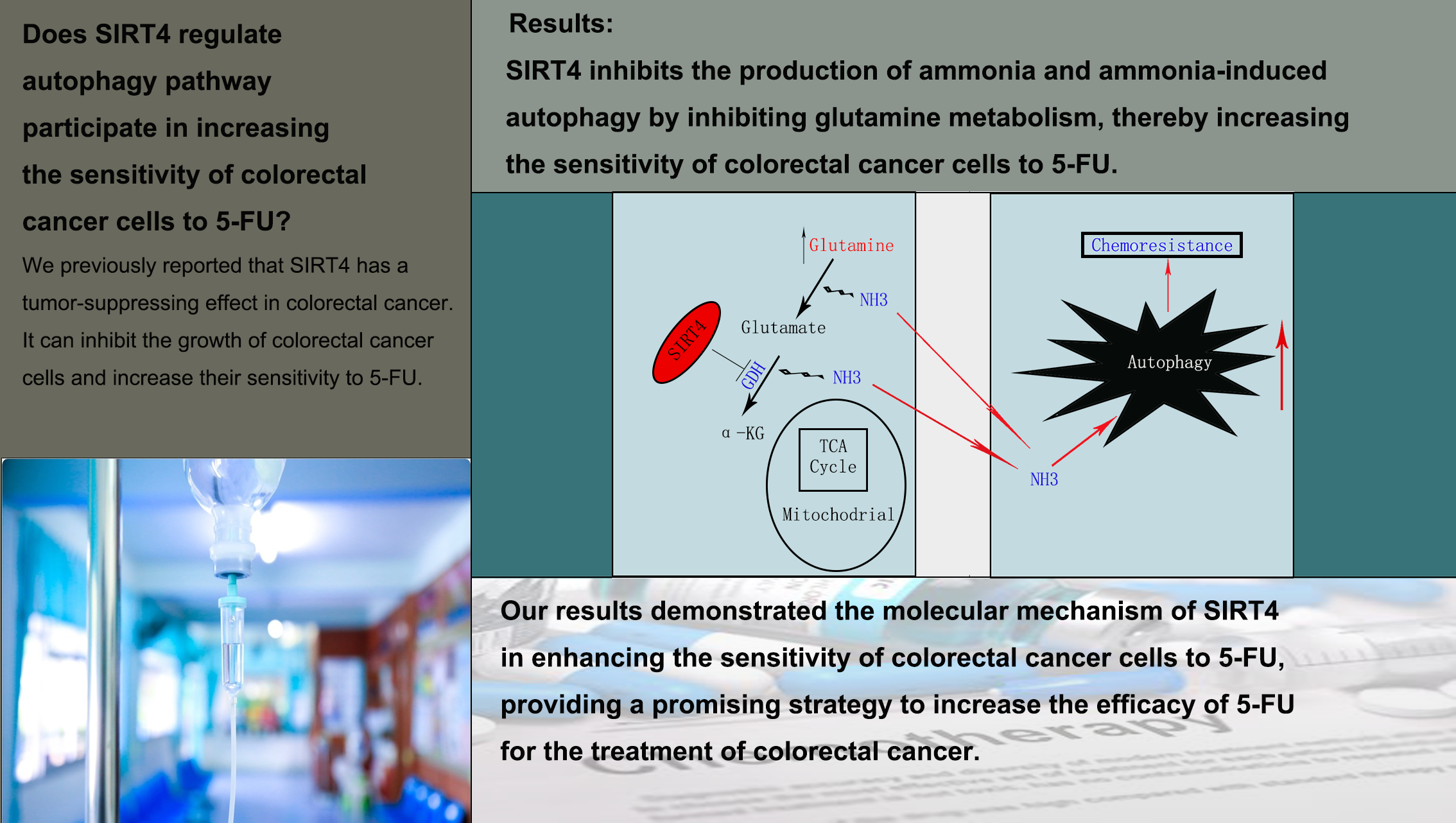
In our previous study, we discovered that SIRT4 plays an important role in the progression of colorectal cancer, and overexpression of SIRT4 enhances the sensitivity of colorectal cancer cells RKO and HT29 to the chemotherapy drug 5-FU[27]. 5-FU is a first-line chemotherapy drug that is widely used in colorectal cancer patients. However, its efficacy is limited by chemotherapy resistance. In this study, we discovered that SIRT4 overexpression enhanced the sensitivity of colorectal cancer cells RKO and HT29 to 5-FU, while SIRT4 interference reduced their sensitivity to 5-FU. In terms of mechanism, we found that SIRT4 inhibited autophagy, which caused chemotherapy sensitivity changes in the colorectal cancer cells. Furthermore, SIRT4 was found to inhibit glutamine metabolism, which in turn inhibited ammonia-mediated autophagy.
According to our results, SIRT4 can improve the chemotherapeutic sensitivity of colorectal cancer cells, which was consistent with previous reports. Zhu et al. found that knocking out SIRT4 decreases the sensitivity of HT29 cells to 5-FU and oxaliplatin[35]. We previously reported that overexpression of SIRT4 increases the inhibition rate of 5-FU on RKO and HT29 cells in vitro[27]. In this study, the regulatory effect of SIRT4 on the sensitivity of colorectal cancer cells to 5-FU was validated by SIRT4 overexpression and interference. Resistance to 5-FU is a significant obstacle that affects the efficacy of colorectal cancer treatment. Many mechanisms have been implicated in the 5-FU resistance process, such as DNA repair defects[36], drug export transporters, and anti-apoptosis[37]. Because autophagy plays a dual role in tumor progression, the relationship between autophagy and tumor resistance has received extensive attention[38, 39]. Many studies have found that inhibiting autophagy improves tumor cell resistance to chemotherapeutics[11, 40–43].
Like many autophagy-related genes, the expression and function of the SIRT family are also affected by intracellular pressure and nutritional conditions[36]. There is currently no evidence of a link between SIRT4 and autophagy in the literature. However, studies have found that knocking out the SIRT1 gene disrupts or eliminates autophagy in mammalian cells[37]. SIRT1 regulates autophagy by deacetylating autophagy-related proteins or modulating the binding sites of transcription factors to the promoter regions of autophagy-related genes[38]. Both SIRT2 and SIRT5 have also been reported to be related to autophagy[39, 41]. Based on LC3 fluorescent particle detection and analysis of the LC3 and p62 expression levels, we discovered that SIRT4 overexpression inhibited autophagy in RKO and HT29 cells, while SIRT4 interference enhanced autophagy. In addition, we examined the effects of SIRT4 on autophagy-related proteins LC3 and p62 by adding the autophagy inhibitor bafilomycin or inducing autophagy by starvation. Our findings revealed that SIRT4 affected autophagic proteins by inhibiting autophagic flux rather than autophagic particle degradation. When bafilomycin was added, the effect of silencing SIRT4, which enhances the sensitivity of RKO and HT29 cells to 5-FU, was not observed. Similarly, the sensitivity of RKO and HT29 cells to 5-FU was also not increased after silencing the autophagy-related gene ATG5 using an adenovirus. These results indicated that SIRT4 affects the sensitivity of RKO and HT29 to 5-FU by inhibiting autophagy.
Many studies have found that ammonia and autophagy are closely related[29, 44–48]. According to recent studies, ammonia has a dual effect on autophagy in vitro, inhibiting autophagy at high concentrations (above 20 mmol/L) but promoting autophagy at low concentrations. At high concentrations, ammonia impairs lysosomal function by increasing lysosomal pH and water influx, which can cause substrate degradation defects and lysosomal swelling[47, 48]. In contrast, ammonia strongly activates autophagy in vitro and in vivo at low concentrations (0.2–10 mmol/L)[29, 45, 49]. Glutamate-amino ligase, also known as glutamine synthetase (GS), catalyzes the conversion of ammonia to glutamine. Besides the urea cycle, it is the other major ammonia detoxification system in the human body[50, 51]. Glutamine is the most abundant free amino acid in the human body that plays an important role in metabolic pathways, cell signal transduction, proliferation, and autophagy. Glutamine-dependent autophagy regulation is linked to cancer cells in particular. It promotes the proliferation of cancer cells by increasing the uptake and decomposition of glutamine as a nitrogen source and decomposition[44, 52]. Interestingly, ammonia released from glutamine metabolism has been reported to activate autophagy [46] strongly. Recent studies have found that SIRT5 can regulate glutamine metabolism to modulate ammonia-induced autophagy and mitochondrial autophagy[41].
In this study, SIRT4 inhibited the ability of RKO and HT29 cells to utilize glutamine as an energy substance. Furthermore, SIRT4 inhibited GDH enzyme activity and protein levels. Moreover, we discovered an interaction between SIRT4 and GDH. Our findings demonstrated that SIRT4 inhibits glutamine metabolism in RKO and HT29 cells by inhibiting GDH. The ammonia concentration in the culture medium was found to be decreased after overexpression of SIRT4. In addition, when glutamine was deprived, or BPTES was added, the effect of SIRT4 on the ammonia concentration in RKO and HT29 cells was not observed. According to these findings, SIRT4 inhibits glutamine metabolism in RKO and HT29 cells by inhibiting GDH, thereby inhibiting ammonia-induced autophagy.
New tumor treatment strategies that block metabolic pathways are currently being developed[53, 54]. For example, glucose inhibitors have been used to target the glucose metabolism pathway in the treatment of tumors[55]. Tumor cells can survive with reduced glucose metabolism by activating additional metabolic pathways, including glutamine metabolism. Mitochondrial glutamine metabolism can compensate for the lack of glucose and supplement the mitochondrial tricarboxylic acid cycle[32]. Therefore, the inhibition of both glucose and glutamine metabolism has great potential for cancer treatment[56]. Overexpression of SIRT4 has been shown to increase the sensitivity of Burkitt's lymphoma cells to glucose deprivation and increase cell death associated with glucose metabolism inhibitor therapy[57]. In this study, we discovered that overexpression of SIRT4 increased the mortality of colorectal cancer cells caused by glucose deprivation. In addition, the combination of SIRT4 overexpression and glucose inhibitor 2-DG significantly increased the mortality of colorectal cancer cells. Therefore, these results demonstrated the therapeutic potential of SIRT4 targeted metabolism, especially when used in combination with glucose metabolism inhibitors to treat tumors.
{kind=link}