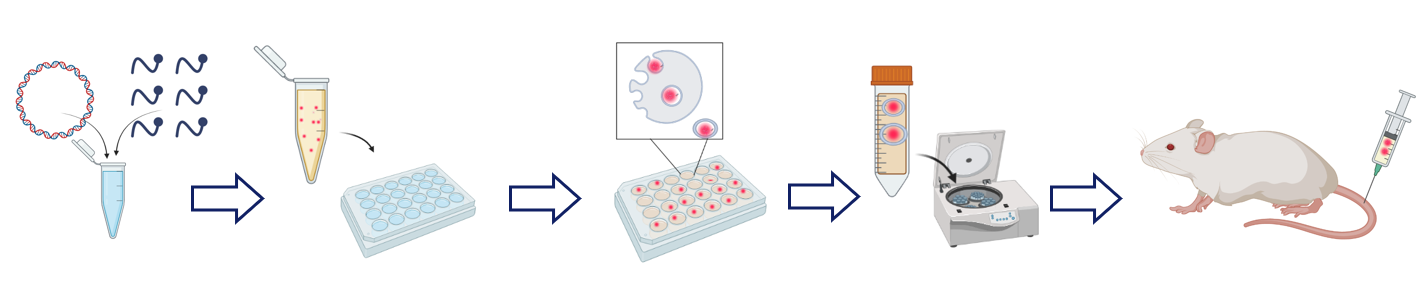
1. Design of proof-of-concept study of EV loading through donor cells transient transfection with pDNA/CPP complexes
We aimed to confirm if pDNA/CPP complexes can be loaded to the excreted EVs. We evaluated the efficacy of the method through quantification of fluorescently- or radio-labeled pDNA in exosomes or EVs. Briefly, cells were incubated with labeled-pDNA/CPP complexes, the transfection media was substituted with fresh media, followed by EV production and conditioned media collection. Thereafter, EVs were isolated by the polymer precipitation method. Samples were analyzed and the proportion of labeled-pDNA in the total exosome or total EV population was measured (Figs. 1a, 1b, and 2a).
1.1 Quantification of Cy5-labeled pDNA in exosomes
pDNA was covalently labeled with Cy5 dye prior to complex formation. Cy5-labeled-pDNA was complexed with CPP PF14 before donor cells transfection. The efficacy of the loading method was assessed in CHO-K1 and N2a cells. The total exosome population was characterized by two different approaches: (1) immune-labeling of isolated EVs with exosome-specific CD63 monoclonal antibody (CD63 mAb) (Fig. 1a); (2) or double transfection of donor cells with two plasmids: model CD63-GFP plasmid and reporter Cy5-labeled plasmid. In the case of efficient double transfection, cells produce exosomes with specific surface-exposed CD63 fused with a fluorescent protein and contained the Cy5-labeled plasmids (Fig. 1b).
In both CHO-K1 and N2a cell lines, we detected a high percentage of labeled-pDNA loading efficacy ranging from 76–86% when cells were co-transfected with CD63-GFP and Cy5-labeled plasmids. When EVs were characterized by CD63 immuno-labeling, the detected loading efficacy was significantly lower compared to double transfection strategy. For the CHO-K1 cell line, it was 42%, and for the N2a cell line 19%. We hypothesized that lower EV loading efficacy in the donor N2a cells compared to the level achieved in CHO-K1 cells can be caused by suboptimal conditions used for donor cell transfection and N2a cells to be more resistant to transient transfection (Fig. 1c).
The difference in loading efficacy measured by the application of two different characterization approaches can be biased due to the protocol used for flow cytometry assay. In the case of immuno-labeling, we evaluate the percentage of cargo-containing exosomes in the total EV population. When exosomes were characterized by reporter protein detection, we evaluate cargo-containing exosomes in total exosomes population with CD63-GFP reporter protein on the membrane surface. We speculate that EVs produced by double-transfected donor cells are more likely to contain Cy5-labeled plasmids compared to the total EV population.
Based on the data from our previous work, we hypothesized that the labeled-pDNA complexation with CPP and incorporation into exosome can lead to a reduction of fluorescence intensity, which could alter the efficacy of reporter Cy5 detection.
To mitigate possible alternation in measurement data caused by method limitations, we applied another approach to the quantification of loaded pDNA to our experimental setup.
1.2 Quantification of radio-labeled pDNA in purified EVs
The pDNA was labeled with [α32P]dCTP and used to form pDNA/CPP complexes (Fig. 2a). Labeled-pDNA was complexed with CPP PF14 before donor cells transfection. The efficacy of the loading method was assessed in CHO-K1 cells. With this assay, we quantified the efficacy of transfection by measuring the amount of cargo radio-labeled-pDNA. At each EV production and extraction step, the samples were collected to evaluate the NA recovery. We observed that approximately 87% of the signal was detected from the transfection media, which reflects the efficacy of donor cells’ overall transfection efficacy. Donor cells collected after the EV production step contained 0.8% of input material, 2.1% was detected in conditioned media prior to purification, and 0.3% of input cargo was recovered in the final purified EV fraction (Fig. 2b). By this quantification method, we measured the proportion of the labeled-pDNA in the final isolated EV population and did not evaluate the ratio of successfully transfected EVs. It should be noted that samples combined make up 95% of input material signal, and ~ 5% was lost during the experiments. Here we concluded that despite the transfection must be optimized, loading of the EVs through transient transfection of donor cells is confirmed.
1.3 Confirmation of proof-of-concept of EV loading through donor cells transient transfection with pDNA/CPP complexes
We observed a significant difference between fluorescently- or radio-labeled pDNA measurements in the total exosome or EV population. The positive signal in the final isolated EV population was 0.3% of total input material compared to 19–86% of the recovery rate in the case of fluorescent-labeled pDNA evaluation (Fig. 1c and 2b). When measuring the proportion of radio-labeled pDNA, we assess pDNA recovery on each step from donor cell transfection to EV purification (Fig. 2b). In the case of fluorescent-labeled pDNA detection, we evaluate the success of cargo-loading of EV, not considering donor cell transfection efficacy (Fig. 1c). When assessing the radio-labeling strategy we observed that ~ 87% of input material was detected during transfection media either been rapidly excreted or not penetrating cells. The donor cells and conditioned media combined contained ~ 3% of input material, purified EVs ~ 0.3%, and about 5% of input material was lost during the experiments. By evaluation of radio-labeled pDNA, we have estimated the percentage of input material loaded into isolated EVs, with no data on the percentage of cargo-loaded particles from the total EV population.
Based on data from labeled-pDNA quantification in exosomes or EVs, we confirmed that this type of vesicle can be loaded with the NA. We hypothesized that with further optimization of the protocol yield of efficiently loaded exosomes can be improved. It should be mentioned that although the detection of the labeled cargo is often used for transfection efficacy evaluation, it does not reflect the bioavailability and bioactivity of the cargo molecules. The detection of a label-positive-EV population is an initial indication of the probability of pDNA/CPP being loaded into the excreted EVs.
2. Transfection of recipient cells with the loaded EV-containing media
The quantification of labeled-pDNA provides an estimate of EV loading efficiency but does not reflect the bioavailability of the pDNA encapsulated in EV. Even if cargo is efficiently loaded into the transport vehicle it can have no effect on recipient cell gene expression and have no therapeutical effect. Therefore, we used pDNA expressing reporter luciferase protein to assess the total reporter levels in recipient cells that can be achieved by using cargo-loaded EVs from donor cells conditioned media23,25,26.
We hypothesized that maximizing the number of successfully transfected cells allows for optimal cargo-loaded EV production23. Most optimal donor cell lines produce a significant amount of clinically relevant cargo containing EVs, optimal CPP efficiently transfects donor cells, and a reasonable fraction of NA is packaged into excreted EVs.
During experimental set-up development, we took into consideration the dependability of transfection efficacy on the used cell line, transfection, and EV production conditions, pDNA dose, CPP features, and charge ratio used for complex formation. Those aspects affect the proportion of loaded EVs and general vesicle production. We performed a series of experiments for broader conditions screening, protocol optimization, and evaluation if conditioned media collected from donor cells can be used for recipient cell transfection (data not shown).
The general workflow for the experimental setup of this section was constant throughout the series. Briefly, donor cells were transfected with pre-formed pDNA/CPP complexes. Next, cells were washed, and fresh media was added to the cells. Followed by the chase period, where donor cells produced cargo-containing EVs and excreted them to media. Afterward, centrifuged conditioned media was added to recipient cells and incubated. The reporter luciferase protein signal was measured from the donor and recipient cell lysate.
2.1 Development of efficient donor cell transfection protocol
We evaluated the transfection efficacy in adherent CHO-K1 and HEK293 cell lines. Complexes were formed with CPP previously shown to be optimal for these particular cell lines, specifically for the CHO-K1 cell line we used CPP PF14, and for HEK293 cell lines complexes were formed with CPPs NF55 and NF71. Transfection of CHO-K1 has shown three logs of lower reporter signal compared to the HEK293 cell line (Supplementary 1). HEK293 cells are originally isolated from human embryonic cells and have more clinical interest compared to CHO-K1, which is a cell line derived from Chinese hamster. Taking this into account, a further HEK293 cell line was used for the protocol optimization.
Transfection of the HEK293 cell line with complexes formed with CPPs NF55 or NF71 resulted in no significant difference in RLU/mg levels of luciferase reporter protein (Supplementary 1). The CPP NF71 has been shown to be an efficient NA transfection vehicle for in vivo applications, and recently we have shown that CPP NF71 has lower toxicity at earlier time points compared to other transfection methods. This provides an excellent starting point for the development of the pDNA-loaded EVs production method and further protocol optimization was performed with CPP NF71.
2.2 Loaded EV production in suspension cells
Next, we hypothesized that the adoption of suspension cells for production has the advantage of decreasing consumption of consumables and improving overall EV yield.
To evaluate if and how the efficacy of donor cell transfection translates to loaded EV production, we transfected donor adherent and suspension HEK293FT cells with the same amount of pDNA. The conditioned media from each cell line was collected, centrifuged, and transferred to recipient cells. Luciferase reporter signal was measured from donor and recipient cell lysate and normalized to total protein. We achieved high reporter signal levels in both suspension and adherent donor cells, where the optimal result was achieved in the suspension cell line up to 1× 1010 RLU/mg. In the recipient cells incubated with conditioned media from suspension donor cells, the reporter expression was only one log lower compared to the signal measured in donor cells (Fig. 3a). Incubation of recipient cells with conditioned media collected from suspension cells led to approximately two logs higher signal when compared to incubation with conditioned media collected from adherent cells (Fig. 3b). This data supports our hypothesis that EV production in suspension culture production of loaded EV with bioavailable pDNA is more effective.
Here we concluded that the production of loaded EVs in suspension cells is more efficient and further loaded EV production is performed in the suspension HEK293FT cell line.
2.3 Scaling up EV production in suspension cell line
The optimized pDNA dosage for a small-scale production cannot always be extrapolated for large-scale production23. Therefore, we optimized conditions for scaling up EV production in the suspension cell line to be translated to industrial production by dosage and transfection time optimization. Results showed no significant differences between the assessed dosages of 3 mg, 4.5 mg, or 6 mg per 6 000 000 cells (Supplementary 2). 500 ng per 1 000 000 cells was selected for further investigation. Next, we optimized transfection, chase period, and pDNA dose variation on loaded EV production (Supplementary 3). Results revealed that in our system 4-hour donor cell transfection period and 4-hour recipient cell incubation with conditioned media are optimal conditions.
3. Isolation and characterization of purified extracellular vesicles
Next, to evaluate EV purification methods, we compared technologies that do not require sophisticated equipment or complex protocols, therefore would be easily used in a standard laboratory setup. Briefly, conditioned media was collected from donor cells and isolation was performed according to the manufacturer's protocol in three different ways: (1) isolation by polymer precipitation, (2) isolation with a tangential flow filter, (3) or isolation by size exclusion chromatography.
With double sandwich ELISA assay, we confirmed that the purified EVs are exosomes and evaluated the concentration of the particles in the sample in ng/µl. With NTA analysis we evaluated the size distribution and concentration of the particles in the sample in particles/ml (Fig. 4). NTA analysis showed a ~ 4–24% EV recovery rate after purification (Fig. 4a). ELISA showed ~ 51–78% exosome recovery rate after purification (Fig. 4b). With both assays, we detected a higher recovery percentage when conditioned media was purified by polymer purification. NTA analysis data reveals a lower recovery rate, compared to ELISA data. It should be noted that NTA describes the particle amount and size distribution of EVs, and ELISA gives insights into exosome concentration in the sample. We hypothesized that assessed purification methods are optimal for exosome purification, eliminating other types of microvesicles.
Here we concluded that EV purification by polymer precipitation was the most efficient in our system and the method was used for further experimental setup.
4. Transfection of recipient cells with pDNA-loaded exosomes
We next aimed to confirm if the purified exosomes transfect recipient cells and if the reporter plasmid is expressed at a reasonable level. Loaded exosomes were produced following a protocol optimized for scaled-up production. Donor cells were transfected with pre-formed pDNA/CPP complexes, following media conditioning. Next, the exosomes from the conditioned media were purified by polymer precipitation and characterized by ELISA, NTA, and total protein amount. For this set of experiments, we included the recipient A549 cell line in the experimental set-up to evaluate the efficacy of loaded EV transfection in more complex conditions as A549 cells are known to be a hard-to-transfect cell line. 8.5×106 of purified exosome particles were used for transfection of 100 000 recipient HEK293FT cells or 50 000 recipient A549 cells. We achieved a high reporter protein signal in the recipient HEK293FT cells up to 3.9×107 RLU/mg, and in the recipient A549 cells up to 4.8×107 RLU/mg (Fig. 5).
Here we conclude that loaded exosomes can successfully deliver cargo-pDNA to the recipient cells in the form where reporter signal protein can be expressed.
5. In vivo transfection with pDNA-loaded exosomes
Next, we were interested in the level of the reporter protein expression in recipient cells in vivo. The pDNA-loaded-EVs were produced by applying two protocols, either CPP PF14 and CHO-K1 cells or CPP NF71 and HEK293FT were used. The purified pDNA-loaded-EVs diluted in PBS, or conditioned media, or pDNA/CPP complexes were administered into 8-week-old female BALB/c mice intravenously via the tail vein with. Postmortem ex vivo tissue analysis revealed that EV-encapsulated pDNA has been expressed in mammalian tissue when administrated intravenously. This affect was achieved only when using CPP PF14 for EV loading with pDNA (Fig. 6a). Administration of unloaded EVs diluted in PBS had lower toxic effect on mice compared to pDNA control. ASAT and ALAT serum marker enzymes were analyzed (Fig. 6b), showing four times lower toxicity level compared to naked pDNA. It should be noted that the mice responded with no visual reaction to EV administration.
{kind=link}