3.3.1 Structure stability and Flexibility analysis:
MD simulations incorporate protein flexibility as well as the role of water, resulting in a more realistic assessment of the protein-ligand complex formation. The simulation trajectory was examined after the MD simulations and the Root-mean square deviation (RMSD), root-mean-square fluctuation (RMSF), and radius of gyration (Rg) values were found to explain the stability of the molecular interactions in the present study. The RMSD of the protein’s backbone atoms represents the global stability of the protein. Therefore, variation in the RMSD with respect to their starting structures throughout MD simulation was used as a metric to assess the stability of protein's backbone atoms. For the qualitative investigation of the stability and convergence of the simulated system, the RMSD of the protein backbone atoms was analyzed (Fig. 1A). Average RMSD values for the last 50 ns of each simulation were reported in Supp. table 5. As expected, some structural variations at the beginning of simulations were observed; however, after 50 ns, all molecular systems show constant RMSD values, indicating system stability for all kinase:inhibitor complexes (Fig. 1A). Because of this, the last 50 ns of each MD simulation were used for cluster analysis to collect the geometries by RMSD, and Figure S3 illustrates the representative structure of the prevalent binding pose of each system. According to Fig. 1A and Supp. table 5, the average RMSD of the last 50 ns of the backbone atoms of AURKA varied from 2.44 Å to 3.42 Å. The average RMSD of the protein without ligand shows the value of 3.01 ± 0.0001 Å. Therefore, the inhibitor binding does not destabilize the overall structure of AURKA, and indeed most ligand-bound systems show smaller average RMSD values than the native apo system.
We also estimated the radius of gyration (Rg) of AURKA at the apo protein level and protein: ligand complex. The average Rg values of the last 50 ns were reported in Supp. table 5, and time evolution plots of these parameters were shown in Fig. 1B. Rg provides an insight into the overall size and dimensions of the protein complexes. It also helps to examine structural drifts in protein complexes. The average Rg values for the apo- and holo-systems (ligand-bound) vary between 19.39 Å to 19.63 Å. Therefore, small, and stable radius of gyration values were observed for all molecular systems (Fig. 1B), confirming the AURKA's compactness and stability in all systems. Overall, the Rg values were similar for all systems and did not change much after inhibitor binding. In addition, ligands remain tightly bound to the binding pocket of AURKA throughout simulation study and their RMSD profiles also suggest that they reached equilibrium (Fig. 1C). The RMSD profile of each ligand, last (50ns of simulation) are given in Supp. table 5. Ligands with a smaller and larger deviation of the atomic positions are the Imatinib (≈ 1.5 Å) and Ponatinib (≈ 3.5 Å). Furthermore, to acquire a measure of the movement of each system relative to the average structure over the whole simulation, the RMSF values were also calculated for the protein by residues (Fig. 1D). The RMSF computed from the MD simulations trajectory reveals that the residual fluctuations over the simulation time for the AURKA protein: ligand complexes, fluctuations were observed at regions of amino acid residues of 146–161, 196–211, 261–276 and 296–311. Higher structural fluctuations were observed in regions known to be involved in ligand binding particularly in catalytic loop regions. The analysis of these values indicated a higher per-residue fluctuation of AURKA upon Vemurafenib and CFI-400945 binding, whilst the smaller values were observed for the AURKA:Imatinib and AURKA:Ponatinib complexes. The C-lobe residues are those that mostly modify their position during all simulations. In addition, except the CFI-400945, the fluctuation of the key interacting residues (Leu139, Val147, Lys162, Tyr212, Gly216, Leu263, and Asp274) upon ligand binding is rather small (< 4 Å). This is particularly evident in the AURKA:Imatinib, probably due to its stable binding/packing and complexation.
3.3.3 Molecular interactions between ligands and AURKA
The best binding poses of ligands obtained from molecular docking and molecular dynamics calculations were thoroughly studied to understand binding interactions of these ligands with amino acid residues of AURKA. Molecular interactions and ligand occupancy of these small molecules in the AURKA binding site of 2J4Z were analyzed. All six molecules showed common hydrophobic contacts with amino residues Leu139, Tyr212, and Gly216. In addition to this, these ligands made molecular interactions through hydrophobic contacts with many residues viz. Gly140, Val147, Ala160, Lys162, Leu194, Leu210, Glu211, Ala213, Pro214, Leu215, Thr217, Gly216, Arg220, Asp256, Glu260, Asn261, Leu263, Ala273, Asp274, Ser278, Val279, His280, and Pro282. A wide range of hydrogen-bonding interactions was also observed with amino acid residues Arg137, Leu139, Glu211, Ala213, Tyr212, Arg220, Arg239, Asn261, Asp274, Val279, and His280. Further, it was noticed that these molecules exhibited binding interactions with residues Val147, Lys161, Glu211, Tyr212, Arg220, Leu263, and His280, which are important for ATP binding in AURKA. A summary of the types of binding interactions between the selected ligands and the interacting amino acid site residues of 2J4Z has been given in Supp. table 8. As expected, all tested ligands interact with some key residues of ATP-binding sites, such as Leu139, Tyr212, Arg220, and Leu263. Precisely, the Van der Waals contacts and hydrophobic interactions with the nonpolar side chains of Leu139, Val147, Ala160, Leu194, and Leu263 appear to further stabilize these ligands in the ATP-binding site. However, CFI-400945 and Vemurafenib (comparable binding affinity) do not interact with Val147 and Ala213 of proteins, which are known to be important for binding in this pocket. This may also contribute to their weak affinity for the protein. Within the hinge region of AURKA, ATP forms crucial hydrogen bond interactions with Glu211 and Ala213 and purine base capture a large hydrophobic pocket created by residues Leu210, Leu139, Val147, Ala160, Leu194, and Leu263. Additionally, the phosphoryl groups of ATP create a hydrogen bond network with residues Lys143, Phe144, Gly145, Lys162, Asn261, and Asp274. Due to the high affinity of ATP for AURKA, our inhibitors would participate with these key interactions and may compete for and displace ATP from the active site. Consistent evidence suggests that the compounds occupying the ATP-binding site and forming hydrogen bonds to Ala213 in the hinge region of AURKA are known to be potential inhibitors [58, 59]. Our study shows Ponatinib, Imatinib, GSK-2334470 and MK2i3 form hydrogen bond interactions with the backbone aminocarbonyl oxygen of Ala213 in the hinge region. Furthermore, in many kinases, gatekeeper mutations that convert threonine gatekeeper residue to a larger hydrophobic residue have been shown to confer drug resistance [60, 61]. Moreover, amino acids like Lys162 and Asp274 are considered kinase-dead mutants. These residues are essential for the proper kinase activity of AURKA. There are reports of known inhibitors of AURKA showing interactions with Val147, Lys162, Leu178, Leu194, Glu211, Tyr212, Ala213, Leu263, Ala273, Asp274. Thus, the simulation studies support our inhibitors that can be used against AURKA. Since Ponatinib has a comparable affinity to Imatinib, a known type II inhibitor of the inactive conformation of kinases [62, 63], it was expected that they could also be a potential inhibitor of AURKA. In agreement with the literature, the binding pocket of Imatinib extends into the allosteric site adjacent to the ATP-binding site of the inactive conformation. This allows the simultaneous interaction with residues of the nucleotide-binding site, the activation loop, and the DFG motif. In addition to this inhibitor, the Ponatinib and GSK-2334470 also interact with residues from the activation loop, the His280, and Pro282, and probably this dually covering of both sites is responsible for the greater binding affinity of these compounds. Asteriti et al., 2017 had investigated competitive inhibitors targeting Aurora kinase activity at the ATP-binding site for therapeutic purposes in osteosarcoma cells [64]. Application of various AURKA inhibitors targeting the ATP binding site and catalytic site of the protein showed an outcome of stable disease in approximately half of the patients [65]. The stability of protein-ligand complex is important in understanding an inhibitory action of small molecule ligands because of their critical role in modulating protein function. Initially, molecular systems of known serine/threonine kinase inhibitors of AURKA were computationally investigated, to find crucial structural determinants of AURKA specific binding, especially the hot spots of interactions, which would aid in designing novel selective AURKA inhibitors for cancer therapy. Based on molecular docking, MD simulations, and subsequent energy calculations, all the protein-ligand complexes in our study possessed stable dynamics with improved stability and enhanced affinity. Binding energy calculations revealed that the VdW forces were the principal interactions that increase net binding energies. Furthermore, our findings validated the previously reported in vitro studies of CFI-400945 inhibiting AURKA [66].
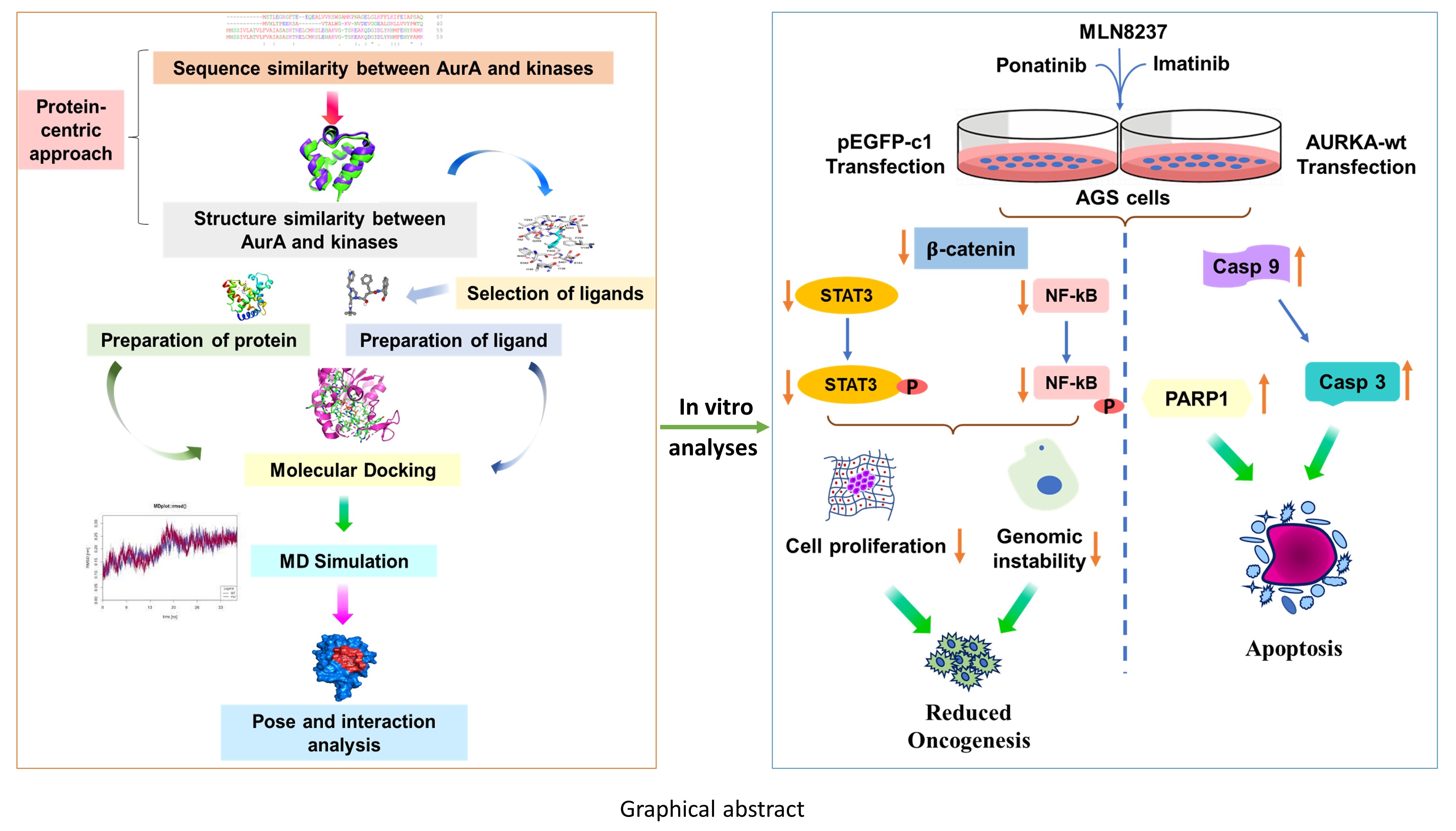{kind=link}