Microarray data
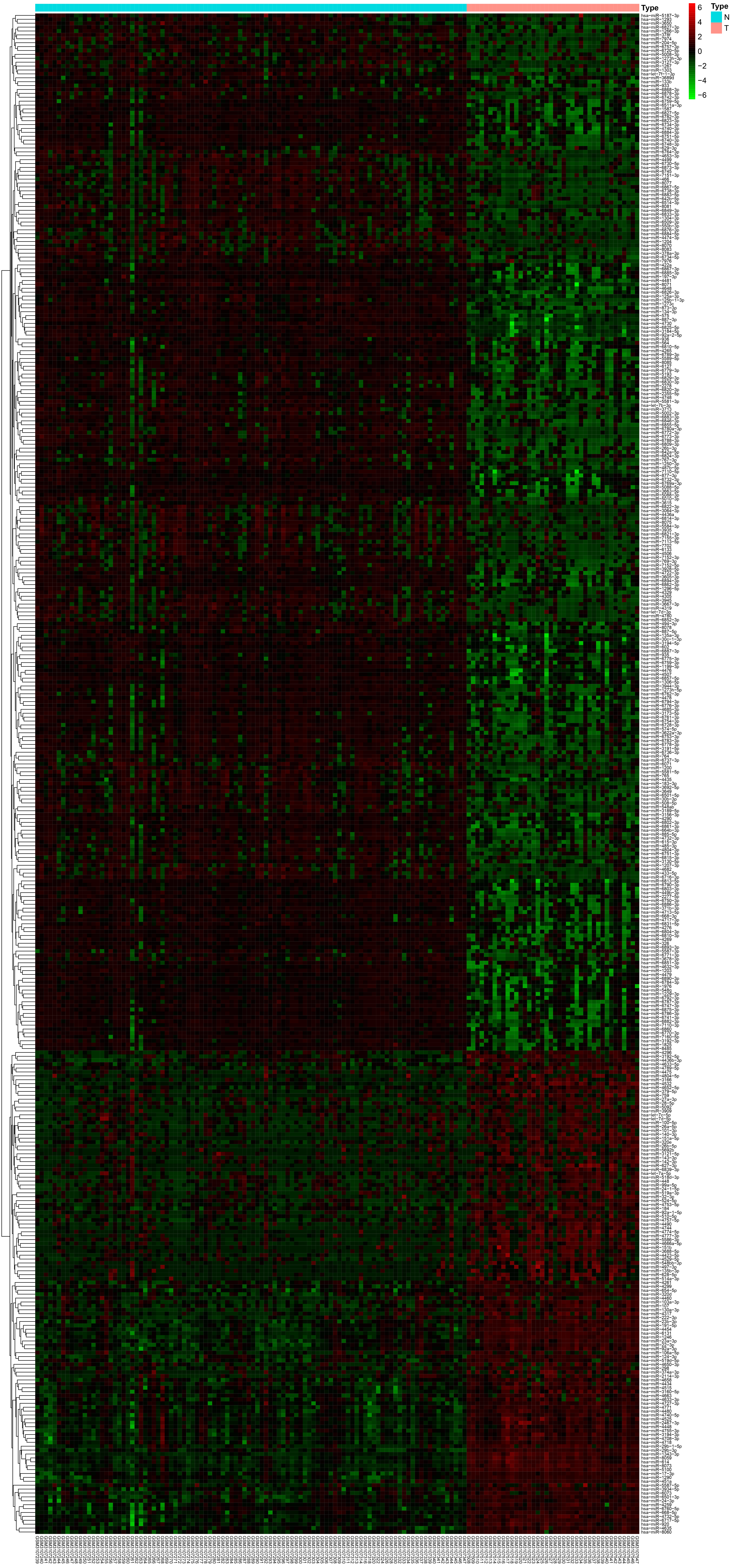
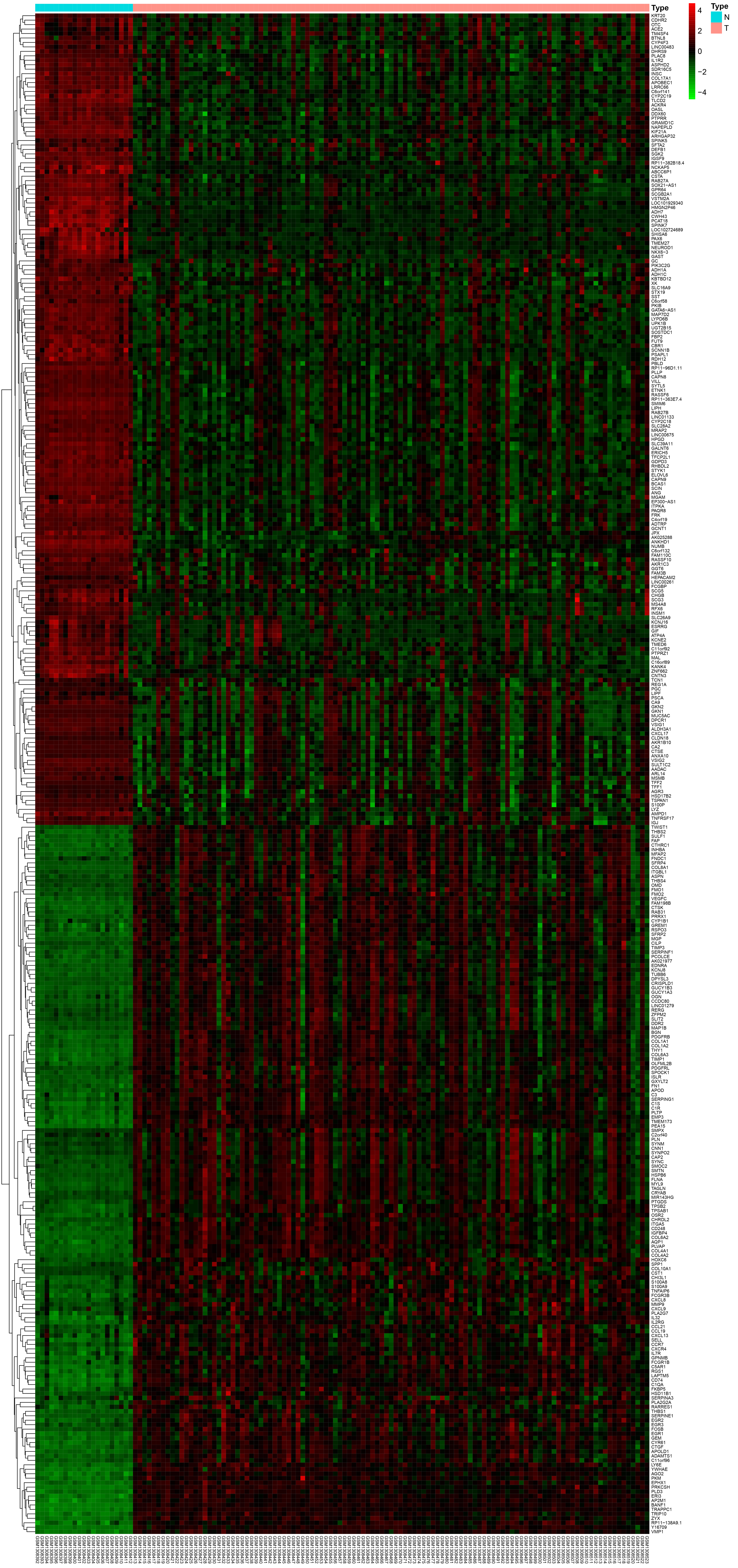
The gene expression profiling dataset (GSE54129) and miRNA expression profiling dataset (GSE113486) were obtained from the GEO database (http://www.ncbi.nlm.nih.gov/geo). The GSE54129 and GSE113486 dataset were based on the GPL10687 platform (Affymetrix Human Genome U133 Plus 2.0 Array, RuiJin Hospital,SJTU) (submission date: Jan 16, 2014) and the GPL21263 platform (3D-Gene Human miRNA V21_1.0.0, Toray Industries Inc.) (submission date: Apr 20, 2018), respectively. The array data of GSE54129 includes 111 gastric cancer samples and 21 normal samples. The array data of GSE113486 includes 40 gastric cancer samples and 100 normal samples.
Data processing
Background correction and standardization were performed on the downloaded original series matrix files and the annotation SOFT tables using Perl 5.26 (http://www.perl.org/). In the case of multiple probes corresponding to a single gene, the gene expression value was determined as the average of the probes. R software (version 3.6.1, https://www.r-project.org/) and the "limma" package [21] were used to identify DEGs and DEMs. Analysis was performed using the "limma" package, and adjusted p-value < 0.05 and | log2FC | > 2 were used as cutoff criteria for the identification of DEGs and DEMs.
Identification of upstream TFs and target genes of DEMs
FunRich [22], which provides functional explanations for a mass of genes (http://www.funrich.org/), was used to identify the ten most significant upstream TFs that regulate DEMs. The target genes of DEMs were predicted using TargetScan 7.2 (http://www.targetscan.org/), which predicts the target genes of miRNAs by finding specific sequence complements for each miRNA seed region.
Establishment of a miRNA-mRNA regulatory network
First, the target genes were combined with DEGs to select DEM-differentially expressed target gene pairs, and a miRNA-mRNA regulatory network was established. Then, we used Cytoscape software (version 3.7.0) to visualize the network. Finally, cytoHubba, a Cytoscape plugin, was used to identify the top ten key molecular markers ranked by "Degree".
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses of DEMs
We used FunRich for GO enrichment analysis and DIANA-mirPath v3.0 [23] to identify the KEGG pathways that were significantly related to DEMs in the regulatory network. For this tool, we chose to use the archives in microT Web Server v5.0 to analyze validated miRNA-gene interactions [24]. The GO analysis includes biological processes, cellular components and molecular functions categories. An adjusted p-value < 0.05 was considered statistically significant.
GO and KEGG pathway enrichment analysis of DEGs
GO and KEGG pathway enrichment analysis was performed using the clusterProfiler package [25]. A p-value < 0.05 was used as a threshold for identifying significant GO terms and pathways. A circle diagram of the GO enrichment results for mRNAs in the miRNA-mRNA interaction network was plotted using the GOplot package.
Hub genes and miRNA analysis
Survival analysis of genes was performed using the online tool Kaplan-Meier Plotter (http://kmplot.com/); survival analysis of miRNAs was performed using the online database OncomiR [26] (http://www.oncomir.org/). A log-rank test p-value < 0.05 was considered statistically significant. The online database Oncomine (http://www.oncomine.com) was used to evaluate the expression of key genes in GC.
Clinical samples and immunohistochemistry assay
The 56 GC specimens and 47 adjacent tissue specimens (at least 5 cm from the tumor edge) were collected. All patients with GC were diagnosed by pathology before surgery and did not undergo chemoradiotherapy. Our study was approved by the Ethical Committee of the Second Affiliated Hospital of Nanchang University and written informed consent was obtained from all patients. The samples were incubated with an anti-ZFPM2 (ab121212, Abcam, 1:100) antibody. The expression of the ZFPM2 protein was assessed according to the degree of staining.
Establishment of GC cells with stable knockdown of ZFPM2
The GES-1, AGS, MKN45, MKN28, BGC803, MGC803, BGC823 and SGC7901 cell lines were cultured in Roswell park memorial institute (RPMI) 1640 medium or Dulbecco's modified Eagle medium (DMEM) at 37 °C and 5% CO2, and the medium was replaced once every 2 to 3 days. All experiments were performed when the cells reached 75%-85% confluence and were in a logarithmic growth phase. SGC7901 and MGC803 cells were transfected with lentivirus-mediated control shRNA or shRNA targeting ZFPM2 and were designated vector and shZFPM2, respectively. The lentiviral-mediated shRNA was obtained from Hanbio Biotechnology (Shanghai, China); the following sequence fragments were inserted into the vector: control shRNA, GACGUAGCCAACCUCAAUAUU; shRNA-1, 5'-GGCUCUGUUUGCACUUUAUUU-3' and shRNA-2, 5'-GCUGCAAGUAUGAAUUUAAUU-3'. At 72 hours post infection, the cells were cultured in medium containing 5 µg/ml puromycin (Sigma, Missouri, USA) until all uninfected cells were killed by puromycin, thus obtaining stably transfected cell lines.
Western blot assay
Stably transfected cells were lysed with RIPA buffer to obtain total protein, and then the protein was transferred to a PVDF membrane after separation by gel electrophoresis. Anti-ZFPM2 (PA5-29094, Invitrogen, 1:3000) was used as the primary antibody. HRP-linked anti-rabbit IgG (ab6721, Abcam, 1:5000) and HRP-linked anti-mouse IgG (ab6709, Abcam, 1:5000) were used as the secondary antibodies. GAPDH (5174T,CST༌1:1000) and actin (8457T༌CST༌1:1000) served as internal standards. The protein quantity was assessed by a BCA assay kit (Beyotime Biotechnology, China).
Quantitative real-time polymerase chain reaction (qRT-PCR) assay
Total RNA was isolated from the samples with TRIzol reagent (Invitrogen), and then total RNA was used to synthesize cDNA using the PrimeScript reverse transcriptase kit (TaKaRa, Shiga, Japan). qRT-PCR was performed to analyze the levels of ZFPM2 mRNA in samples. The primer sequences for amplification were as follows: ZFPM2-forward (5'-TTGCTCATCTCCGAACGTGAA-3'), ZFPM2-reverse (5'-CGCAGCTCAGATTTTCAGGC-3'), GAPDH-forward (5′-CTTTGGTATCGTGGAAGGACTC-3′), and GAPDH-reverse (5′-AGTAGAGGCAGGGATGATGT-3′). The mRNA expression was analyzed using the 2−ΔΔCT method.
Cell proliferation assay
SGC7901 and MGC803 cells in the vector/shZFPM2 groups were inoculated into 6-well plates. When the cell density reached 75%-85%, we used a pipette tip to make a 0.5 cm line in the middle of the well. The cells were then cultured with serum-free medium, and placed in a 37 °C and 5% CO2 incubator. Next, we obtained images at 0 hours, 24 hours, and 48 hours to observe the wound healing process. Additionally, when the cell density reached 75%-85%, the cells were labeled with EDU, fixed with 4% paraformaldehyde, decolorized with glycine solution, incubated with 0.3% Triton X-100 PBS, and then sequentially stained with Apollo and Hoechst. After staining, laser scanning confocal microscopy observation and imaging were performed (Hoechst 33342 is blue light). An anchorage-independent colony formation assay was performed using soft agar growth of 3 × 10 3 cells for 10–14 days.
Cell migration and invasion assays
Precooled Matrigel was added to the bottom of the Transwell chamber to coat it with Matrigel. It was then placed in an incubator until Matrigel solidified. SGC7901 and MGC803 cells (control group and shZFPM2 group) were resuspended and seeded into the upper chamber in serum-free medium at a density of 2 × 105/ml, and the lower chamber was filled with 20% FBS medium. After 24 hours of incubation, the non-migrated cells in the upper chamber were wiped off with a sterile cotton swab. Finally, the migratied cells were examined by crystal violet (0.1%) staining and counted under a microscope. Migration experiments did not include Matrigel.
Statistical analysis
The t test was used for comparison between the two groups. A p-value < 0.05 was considered statistically significant. All experiments were repeated three or more times. Data analysis was performed using SPSS 22.0 for Windows (SPSS Inc., Chicago, IL, USA).
{kind=link}
{kind=link}