Materials
Chemicals
Reagent grade 5-chloroisatin, and 2,4-dinitrophenylhydrazine were purchased from Sigma Aldrich. Ethanol as solvent and concentrated acetic acid were used as purchased.
Physical measurements
Elemental analyses were performed with a Thermo Flash EA-1112 series CHNS-O Elemental Analyser. The melting points were deter- mined with a Stuart SMP11 instrument in sealed capillary and are uncorrected. Infrared spectra were obtained (KBr 400-4000 cm-1) on ALPHA FT-IR Spectrometer from Bruker. UV-visible spectra were carried out with GENESYS 10S UV-Vis spectrophotometer. A Bruker AV 400 MHz Spectrometer was used for the 1H and 13C NMR analysis. Mass spectra were obtained on JEOL Gemate II and Autoflex spectrometers from Bruker.
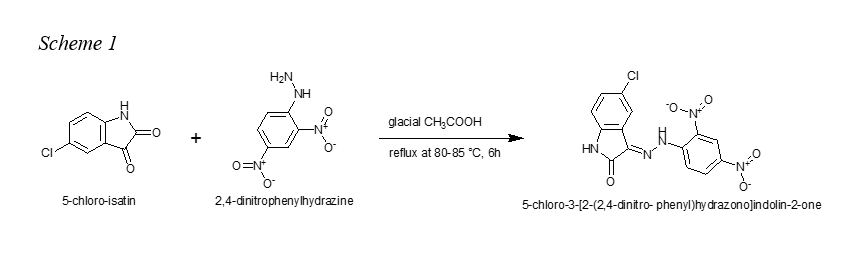
General procedure for synthesis of 5-chloro-3-[2-(2,4-dinitro- phenyl)hydrazono]indolin-2-one (H2L).
To a 200 mL ethanolic solution of 5-chloro-isatin (1.5 g, 8.28 mmol) and 2,4-dinitrophenylhydrazine (1.64 g, 8.28 mmol) was added a catalytic amount of concentrated glacial acetic acid (three drops) under reflux at 80-85 °C (see scheme 1). The resulting solution was further stirred for 6h. After completion of the reaction, the orange-reddish precipitate obtained after cooling overnight was filtered and washed with methanol (100 mL x 2) and dried. Yield 63%; mp > 350 °C; 1H NMR (400 MHz, DMSO‑d6) δ ppm 11.69 (s, 1H), 11.05 (s, 1H), 8.92 (d, J = 2.6 Hz, 1H), 8.61 (dd, J = 9.4, 2.6 Hz, 1H), 8.14 (d, J = 9.4 Hz, 1H), 7.91 (s, 1H), 8.61 (dd, J = 9.4, 2.6 Hz, 1H), 6.98 (d, J = 8.3 Hz, 1H). 13C NMR (100 MHz, DMSO‑d6) δ ppm 164.36, 143.99, 143.06, 140.59, 138.08, 133.06, 132.94, 130.99, 126.29, 124.24, 122.98, 117.36, 116.92, 113.07. FTIR (max/ cm-1): 3372w, 3336w, 3188br, (OH, NH), 3104w, 3057w, 17298.5m (C=O), 16921.5m (C=N), 1612s, 1579s, 1497s (NO2), 1470s, 1449m, 1338s (NO2), 1307s, 1269s, 1230m, 1177s, 1135m, 1109s, 1046m, 847m, 830s, 796m (C-Cl), 719m. Elemental analysis (%): Found: C, 46.40; H, 2.2; N, 19.3 (M+, 361) C14H8ClN5O5; Calcd (%): C, 46.55; H, 2.2; N, 19.0. UV-vis: max (DMSO/nm) 271, 391, 420sh, 560. ESI (methanol) m/z =362 (M+, 30 %), 360 (100, M - 2H), 307 (5, M - CO, - HCN).
AlphaScreen binding assays
AlphaScreen assays were performed as described previously (Tietjen et al. 2021). For RBD-ACE2 assays, 2 nM of ACE2-Fc (Sino Biological, Chesterbrook, PA, USA) was incubated with 5 nM HIS-tagged SARS-CoV-2 Spike-RBDs representing ancestral (“Wild-type” (WT)), beta, delta, lambda, or omicron sequences (SinoBiological) in the presence of 5 μg/mL nickel chelate donor bead in a total of 10 μL of 20 mM Tris (pH 7.4), 150 mM KCl, and 0.05% CHAPS. Test compounds were diluted to 100x final concentration in DMSO. 5 μL of ACE2-Fc/Protein A acceptor bead was first added to the reaction, followed by 100 nL test compound and then 5 μL of RBD-HIS/Nickel chelate donor beads. All conditions were performed in duplicate. Following incubation at room temperature for 2 hours, luminescence signals were measured using a ClarioStar plate reader (BMC Labtech, Cary, NC, USA). Data were then normalised to percent inhibition, where 100% equalled the AlphaScreen signal in the absence of RBD-HIS, and 0% denoted AlphaScreen signal in the presence of both protein and DMSO vehicle control. To measure PD-1 – PD-L1 binding, 0.5 nM of human PD-L1-Fc (Sino Biological) was incubated with 5 nM HIS-tagged human PD-1 (Sino Biological) in the presence of 5 μg/mL protein A and 5 μg/mL nickel chelate donor beads in a total volume of 10 μL of 20 mM HEPES (pH 7.4), 150 mM NaCl, and 0.005% Tween. Proteins and test agents were then added, incubated, and analysed as described above.
Selection of Crystal Structure of Spike/ACE2 Receptor
At the time of this study, four three dimensional (3D) structures of spike/ACE2 complex of SARS-CoV-2 were available from Protein Data Bank (PDB) (Berman et al. 2000; Burley et al. 2017; Burley et al. 2018) and had been solved via X-ray crystallography (PDB codes: 6M0J, 6VW1, 6M17 and 6LZG). The crystal structure 6M0J (Lan et al. 2020) was chosen due to high-resolution and domain completeness. The crystal structure of the Spike RBD/ACE2 complex has 832 amino acid residues divided into two chains (A and E). Chain A is the N-terminal peptidase domain of ACE2 which has 603 residues, while Chain E is the receptor binding domain of the Spike protein from SARS-CoV-2 and has 229 amino acids residues. The structure also had bound metallic cofactors (Zn2+ and Cl-), N-Acetyl glucosamine (NAG), and water molecules.
Molecular Docking Procedures
Generally, molecular docking procedures were performed using similar methods as reported in our previous published papers (Simoben et al. 2018, 2021; Divsalar et al. 2020).
Ligand Preparation
The 3D structure of H2L was generated using Molecular Operating Environment (MOE, Chemical Computing Group 2017). The ligand was prepared for docking using the LigPrep tool, as implemented in the Schrödinger’s software (Schrödinger 2017), where all possible tautomeric forms were generated. They were subsequently energy-minimised using the integrated Optimised Potentials for Liquid Simulations (OPLS_2005) force field (Banks et al. 2005). Finally, 60 conformers were calculated with ConfGen using the default settings and allowing minimization of the output conformations (Watts et al. 2010).
Protein Preparation
The crystal structures of spike/ACE2 complex of SARS-CoV-2 (PDB ID: 6M0J) which is the Wuhan variant, along with the human PD-1/PD_L1 (PDB ID: 4ZQK) were downloaded from the Protein Data Bank (PDB; www.rcsb.org) (Berman et al. 2000; Burley et al. 2017; Burley et al. 2018). All water molecules were deleted using MOE software.38 Further preparations of the protein structures preparation were done using the Protein Preparation Wizard of Schrödinger software (Schrödinger 2017; Sastry et al. 2013). At this stage, bond orders were assigned and hydrogen atoms added, missing side chains were filled using PRIME, and the H-bond network was subsequently optimised. The protonation states at pH 7.0 were predicted using the Epik-tool in the Maestro package commercialized by Schrödinger (Schrödinger 2017; Shelley et al. 2007). The structures were finally subjected to a restrained energy minimization step (rmsd of the atom displacement for terminating the minimization was 0.3 Å) using the OPLS2005 force field (Banks et al. 2005). Furthermore, the different variants/mutants of the spike/ACE complex of SARS-CoV were obtained from the Wuhan 6M0J structure (as mentioned above) by mutation (manual replacement of the residues of interest around the spike receptor binding domain (spike-RBD) using the protein builder module in MOE) in the spike protein sequence. Table 1 shows the various mutations carried out on the Wuhan strain or the wild type (WT) spike RBD/ACE2 to derive the various mutants (β, δ, λ and ο).
Table 1. Mutations manually carried out on the SARS-CoV-2 Wuhan strain (WT) spike RBD/ACE2 to derive the respective mutants.
|
Beta
|
Delta
|
Lambda
|
Omicron
|
|
K417N, E484K, N501Y
|
L452R, T478K, E484Q
|
L452Q, F490S
|
G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, Y505H
|
Docking towards the SARS-CoV-2 Spike RBD/ACE2 and the Human PD_1/PD_L1
Docking procedures were performed using the Glide program in a similar way as previously demonstrated (Simoben et al. 2018, 2021; Divsalar et al. 2020). In this work three grid boxes for the SARS-CoV-2 viral protein RBD/ACE2 human receptor (PDB ID: 6M0J) and one grid box for the human protein complex PD_1/PD_L1 (PDB ID: 4ZQK, Zak et al. 2015) were generated and using specific protein residues. For the ACE2/SARS-CoV-2 protein (PDB ID: 6M0J), the first grid box of interest constituted of the following amino acid residues D93, Q80, Q68, D277, N272, L125, Y32, K523, F494 and N560 around the ACE2 binding site (further shown and discussed in the Results Section). The second choice was around the spike RBD-ACE2 binding site and was generated using the centroid of the following the residues Q771, Y718, N752, P744, M365, A3769, E05, E311. The last avenue to investigate was where the compounds will preferably bind when the whole structure is explored for the generation of a grid. For this purpose, the following amino acids D597, T598, K516, V321, Q121, K578, A283, S91, N746, Q68, P744, E518 and T610 were used to generate a third grid around the ACE2/SARS-CoV-2 protein. On the other hand, the grid box for the PD_1/PD_L1 structure was generated using the residues F63, V63, N66, Y68, E84, L122, E136, I134 and I126; as reported in literature (Horita et al. 2016; Tang and Kim 2019). For all the generated grid boxes, the sides were set to 36 Å. The generated 3D conformers of the prepared ligand were docked into the different receptor grid files. For the docking process, default settings were used with exception of input ring conformation as well as writing a total of 10 poses per ligand conformer from the 20 poses that were included for each ligand conformer. The GlideScore Standard Precision (SP) mode was used as the scoring function (Halgren et al. 2004).
{kind=link}