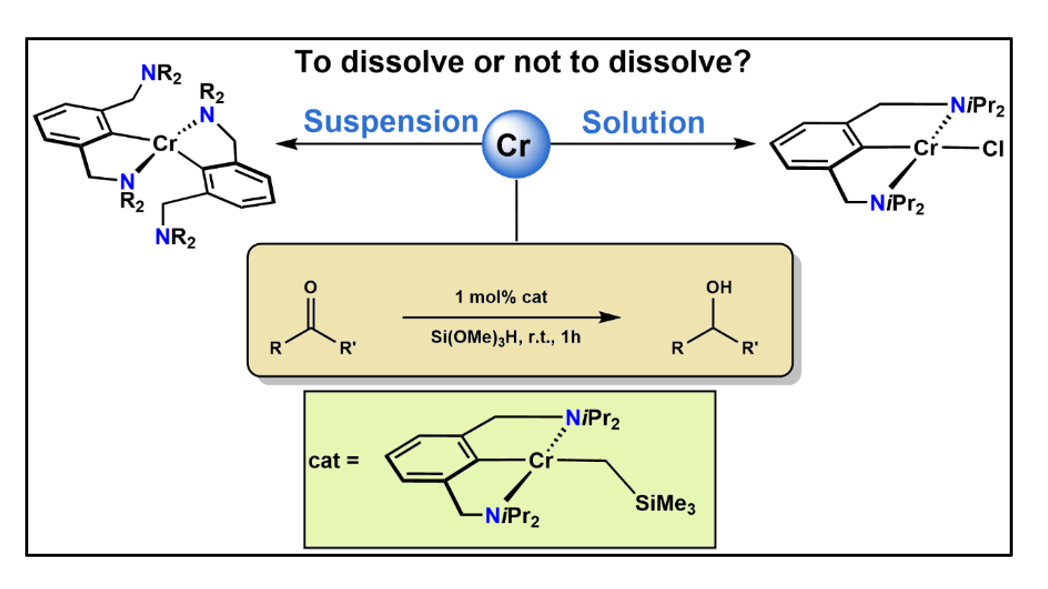
The synthesis, characterization and reactivity of several new Cr(II) and Cr(III) complexes featuring an NCN pincer ligand with an arene backbone connected to amine donors NEt2 and NiPr2via CH2-linkers is described. Reacting the in situ lithiated ligand precursor N(C-Br)NCH2-Et (1) with [CrCl3(THF)3] resulted in the formation of the Cr(III) complex trans-[Cr(κ3NCN-NCNCH2-Et)(Cl)2(THF)] (3). Upon reaction of lithiated N(C-Br)NCH2-iPr (2) with a suspension of anhydrous CrCl2, the Cr(II) complex [Cr(κ2NC-NCNCH2-iPr)2] (4) is formed featuring two NCN ligands bound in κ2NC-fashion. In contrast, when lithiated 2 is reacted with a homogeneous solution of anhydrous CrX2 (X = Cl, Br), complexes [Cr(κ3NCN-NCNCH2-iPr)X] (5a,b) are obtained. Treatment of 5a with 1 equiv of PhCH2MgCl and LiCH2SiMe3 afforded the alkyl complexes [Cr(κ3NCN-NCNCH2-iPr)(CH2Ph)] (6) and [Cr(κ3NCN-NCNCH2-iPr)(CH2SiMe3)] (7). All Cr(II) complexes exhibit effective magnetic moments in the range of 4.7–4.9 µB which is indicative for d4 high spin systems. If a solution of lithiated 2 is treated with CrCl2, followed by addition of an excess of Na[HB(Et)3], the dimeric complex [Cr(κ2NC-NCNCH2-iPr)(µ2-H)]2 (8) is obtained bearing two bridging hydride ligands. [Cr(κ3NCN-NCNCH2-iPr)(CH2SiMe3)] (7) turned out to be catalytically active for the hydrosilylation of ketones at room temperature with a catalyst loading of 1 mol%. X-ray structures of all complexes are presented.
Research Article
Cr(II) and Cr(III) NCN pincer complexes: Synthesis, structure, and catalytic reactivity
https://doi.org/10.21203/rs.3.rs-3224008/v1
This work is licensed under a CC BY 4.0 License
Journal Publication
published 10 Oct, 2023
Read the published version in Monatshefte für Chemie - Chemical Monthly →
You are reading this latest preprint version
Pincer Complexes
Chromium
Hydrosilylation
Silanes
Ketones
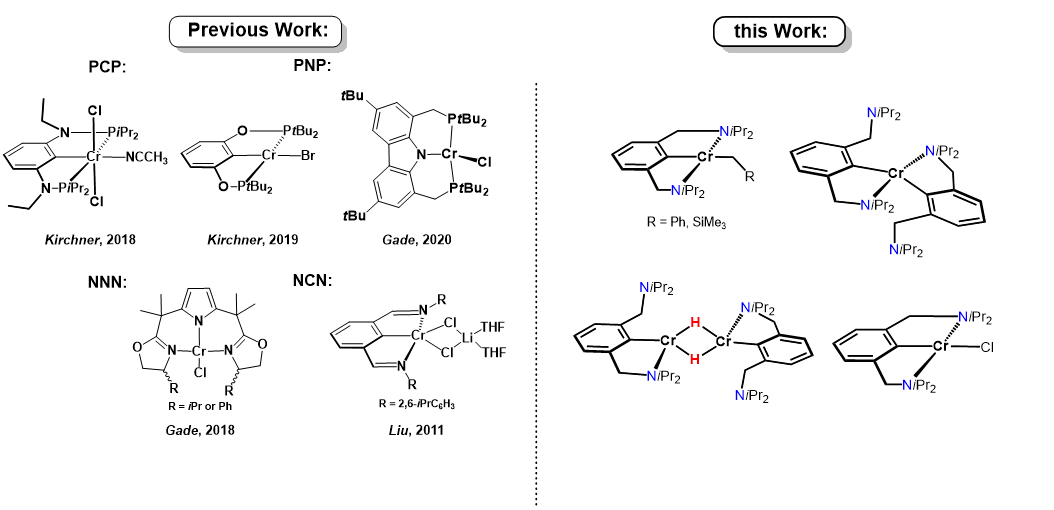
In contrast to the chemistry of transition metal PCP pincer complexes [1–12] which feature an aromatic anionic arene backbone connected to phosphine donors via various linkers (CH2, O, NH, NR), the chemistry of NCN pincer complexes featuring amine donors instead is rich but largely limited to Ni, Pd, and Pt [13–19]. Most notably, van Koten and coworkers, prepared numerous Pd and Pt complexes for applications in catalysis, sensor systems or even as building blocks for biomolecular and peptide chemistry. The major difference affecting the coordination chemistry of NCN ligands, is that the N-atom is significantly smaller than the corresponding P-atom in PCP ligands and the aliphatic NR2 group acts exclusively as a σ-donor. Moreover, NCN ligands are coordinated typically in planar tridentate mer-fashion, but in some cases also a fac geometry was observed [2g].
Expanding on previous work with cobalt NCN pincer complexes [20] we envisioned a further contribution to the virtually non-existing field of chromium NCN pincer chemistry. It has to be noted that some Cr(III), Cr(II) and Cr(I)) PCP pincer complexes were reported recently [21–23] Moreover, Cr(II) complexes containing monoanionic PNP and NNN-pincer type ligands featuring a pyrrole backbone reported by Gade and coworkers (Scheme 1) [24, 25] Gao, Mu and coworkers [26] described the synthesis of Cr(III) bis(imino)aryl NCN pincer complexes which were applied as catalyst for isoprene polymerization.
Herein we report on the synthesis, characterization and reactivity of several new Cr(II) and Cr(III) complexes featuring an NCN pincer with an arene backbone connected to amine donors NEt2 and NiPr2via CH2-linkers. Representative X-Ray structures, EPR-spectra, and DFT calculations are presented.
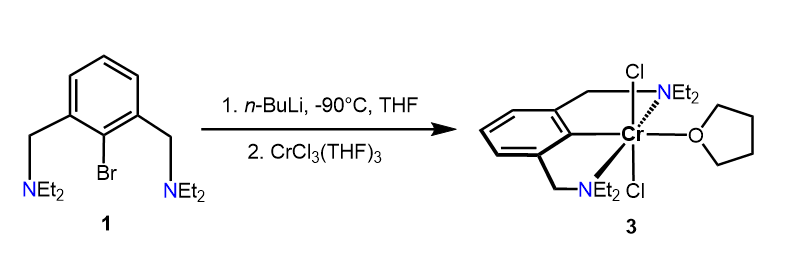
The in situ lithiation of N(C-Br)NCH2-Et (1) with n-BuLi in THF at -90oC followed by addition of [CrCl3(THF)3] resulted, after workup, in the formation of complex trans-[Cr(κ3NCN-NCNCH2-Et)(Cl)2(THF)] (3) in isolated 63% yield (Scheme 2). The measurement of the solution magnetic properties (Evans method, benzene) revealed an effective magnetic moment of 3.7(2) µB which is consistent with three unpaired electrons as expected for a d3 configuration and an oxidation state of + III. Figure 1 depicts the EPR spectrum of 3 obtained from a frozen toluene solution at 100 K, which confirms the expected quartet ground state. This spectrum has been successfully simulated using the following parameters: gx = 1.989, gy = 1.764, gz 2.392 (giso = 2.048) and 53Cr hyperfine coupling constants Ax,1 =82.575, Ay,1 = 0.009, Az,1 = 315.588 and Ax,2 = 19.677, Ay,2 = 251.114 and Az,2 =107.022. The observed effective giso-value is typical for an electronic spin of S = 3/2.
In order to unequivocally establish the ligand arrangement and geometry, single crystals were grown by slow diffusion of n-pentane into a saturated THF at room temperature. A view of the molecular structure is depicted in Fig. 2 with selected metrical parameters reported in captions. The complex adopts a distorted octahedral geometry. While the C20-Cr1-O1 and Cl1-Cr1-Cl2 angles deviate only slightly from linearity being 179.5(4)° and 171.5(4)° respectively, whereas the N1-Cr1-N2 angle deviate significantly from 180o being 155.1(3)°. Similar geometries were recently found for the analogous complexes trans-[Cr(κ3PCP-POCOP-iPr)(Br)2(THF)], trans-[Cr(κ3PCP-PCPNEt-iPr)(Cl)2(THF)] and trans-[Cr(κ3PCP-PCPCH2-iPr)(Br)2(CH3CN)] [21, 23] The Cr-Cipso distance is 1.995(7) Å being comparable to those of other Cr(III) PCP pincer complexes. The Cr-Cipso bond distance in [Cr(κ3PCP-POCOP-tBu)(Br)], trans-[Cr(κ3PCP-POCOP-iPr)(Br)2(THF)], [Cr(κ3PCP-POCOP-tBu)(κ2-BH4)], [Cr(κ3PCP-POCOP-tBu)(NO)(κ2-BH4)], [Cr(κ3PCP-POCOP-tBu)(NO)(Br)] [22], and trans-[Cr(κ3PCP-PCPCH2-iPr)(Br)2(CH3CN)] [23] are 2.084(3), 2.067(5), 2.076(2), 2.068(3), 2.056(2), and 2.052(1) Å, respectively.
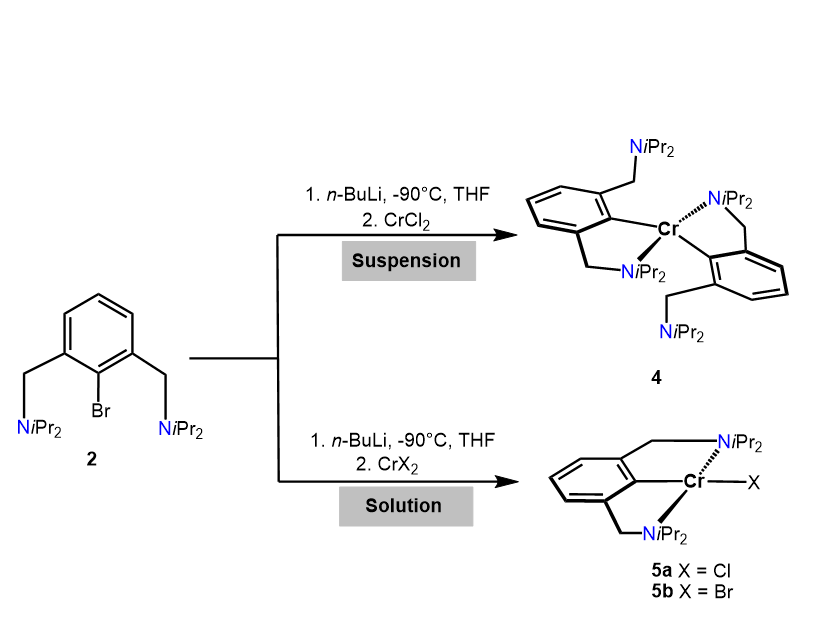
Treatment of lithiated N(C-Br)NCH2-Et (1) with anhydrous CrCl2 resulted in the formation of intractable paramagnetic products. Gratifyingly, the reaction of the bulkier ligand N(C-Br)NCH2-iPr (2) with a suspension of anhydrous CrCl2 under otherwise similar reaction conditions afforded, after workup, [Cr(κ2NC-NCNCH2-iPr)2] (4) in 37% isolated yield (Scheme 3). This complex contains two NCN ligands bound in κ2NC-fashion. Evaluation of the solution magnetic properties of 4 (Evans Method, benzene) showed an effective magnetic moment of 4.7(3) µB which is indicative for a d4 high spin system featuring four unpaired electrons.
Cooling of a saturated n-pentane solution of 4 to -30°C led to formation crystals suitable for single crystal X-ray diffraction studies. A structural view of 4 is depicted in Fig. 3 with selected bond distances and angles given in the caption. Complex 4 adopts a strongly distorted tetrahedral coordination geometry as also seen from the structural parameters τ4 and τ4’ being 0.5051 and 0.5013 respectively (τ4 = τ4’ = 0 indicates an ideal square planar structure, τ4 = τ4’ = 1 indicates an ideal tetrahedral structure and a τ4 ≈ 0.43 and τ4’≈ 0.24 represents a seesaw geometry) [27]. The NCN ligand is coordinated in κ2NC-fashion and features one pendant amine arm.
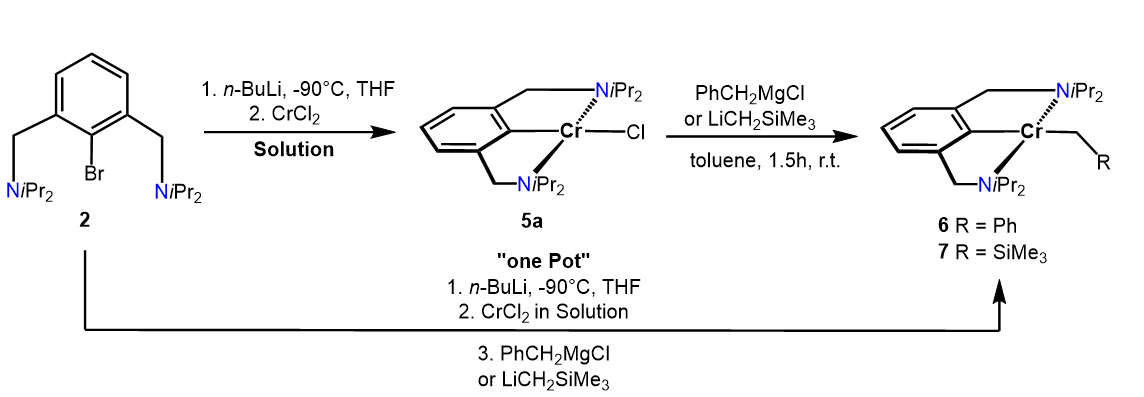
In contrast, when the lithiated ligand N(C-Br)NCH2-iPr (2) is reacted with a homogeneous solution rather than a suspension of anhydrous CrCl2 or CrBr2 in THF, obtained after ultrasonic irradiation, complex [Cr(κ3NCN-NCNCH2-iPr)Cl] (5a) and [Cr(κ3NCN-NCNCH2-iPr)Br] (5b) were obtained in 73 and 75% isolated yields (Scheme 3). As indicated by solution magnetic susceptibility measurements (Evans method, benzene), these compounds are high-spin complexes with a solution effective magnetic moment of 4.8(2) and 4.7(2) µB. This is in agreement with a high-spin d4 center (four unpaired electron) and is in the same range as the theoretical spin-only value of 4.90 µB.
The molecular structure of 5a shows the metal in a typical distorted-square planar configuration (τ4 = 0.1603, τ4’ = 0.0985) [27]. The C1-Cr1-Cl1 angle deviates slightly from linearity 178.59(7) o. The N1-Cr1-N2 angle is 158.52(8) o. The Cr-Cipso bond distance of 2.011(2) Å is comparable to those of other Cr(II) pincer complexes [22].
Treatment of 5a with 1 equiv of PhCH2MgCl and LiCH2SiMe3 in toluene for 1.5 h afforded the alkyl complex [Cr(κ3NCN-NCNCH2-iPr)(CH2Ph)] (6) and [Cr(κ3NCN-NCNCH2-iPr)(CH2SiMe3)] (7), respectively, in 72 and 68% isolated yields (Scheme 4). It has to be noted that complexes 6 and 7 can also be obtained directly from 2 via a one pot reaction (see Experimental Section). Solution magnetic susceptibility measurements (Evans method, benzene) show that these complexes are also d4-high-spin complexes with effective magnetic moments of 4.9(1) and 4.8(1) µB. A structural view of 6 is shown in Fig. 5 with selected bond distances and angles reported in the caption. The coordination geometry around the chromium center is best described by a slightly distorted square-planar arrangement. The C1-Cr1-C21 angle deviates from linearity being 173.29(5) o. The N1-Cr1-N2 angle is 154.99(4)o. The Cr-Cipso and the Cr-Calkyl bond distances are 2.043(1) and 2.249(1) Å, respectively.
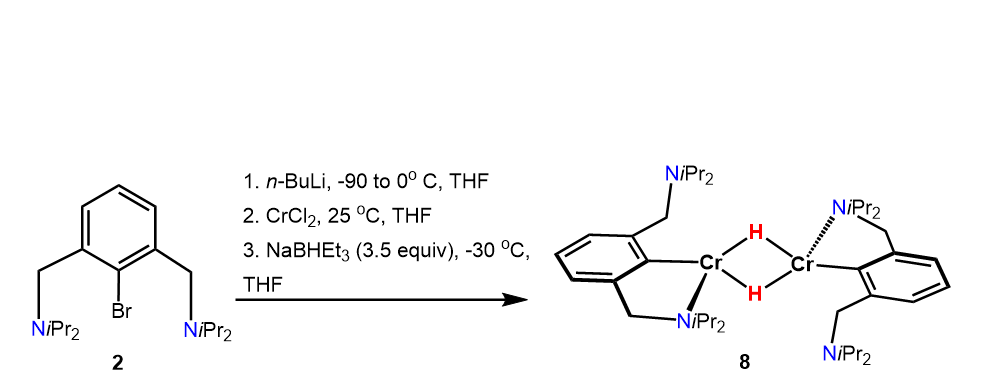
If a solution of lithiated N(C-Br)NCH2-iPr (2) is treated with anhydrous CrCl2, followed by addition of an excess of Na[HB(Et)3] (3.5 equiv, 1M solution in THF) at
-30°C, after work up, the dimeric complex [Cr(κ2NC-NCNCH2-iPr)(µ2-H)]2 (8) is obtained in 35% isolated yield (Scheme 5). A solution magnetic moment of µeff = 6.9(3) µB (Evans method, benzene) was determined which is consistent with eight unpaired electrons as expected for a high-spin d4 configuration of the two metal centers. This also suggest that complex 8 contains no metal-metal bond (vide infra).
The molecular structure of 8 was unequivocally determined by X-ray crystallography. A structural view of 8 is given in Fig. 6 with selected bond distances and angles given in the caption. Complex 8 contains two µ2-hydride ligands bridging the two Cr(II) centers. The distance of Cr1 to its image by inversion symmetry is 2.6941(6) Å which is in line with structurally related compounds found in literature [24–26].
The relevant orbitals obtained by DFT calculations for complex 8 are depicted in Fig. 7 and are consistent with two high-spin d4 Cr(II) centers. The spin density is essentially centered on the metals. The relevant Wiberg index (WI) [28] for the Cr⋅⋅⋅Cr interaction is 0.15 indicating essentially no Cr-Cr metal bond. The two µ2-hydrogen atoms are of hydridic nature with NPA charges of -0.35.
Since a Cr(II) PCP alkyl complex was recently shown to be catalytically active for the hydrosilylation of ketones [22], we investigated the potential the Cr(II) NCN alkyl complex 7 as catalyst for this transformation. Complex 7 (1 mol % based on ketones) was reacted with both aromatic and aliphatic ketones and Si(OMe)3H (2 equiv) in toluene (3 cm3) at 25°C. After workup with K2CO3/MeOH, all alcohols were isolated in yields up to 89% and were characterized by 1H, 13C{1H} and 19F{1H} NMR spectroscopy. These results are summarized in Table 1. The protocol tolerates halide (6a, 6b), ether (6c-6e) and thiophene (6h) functionalities. Lower or no conversion was observed in the presence of amine (6h) and hydroxy (6k) groups. Likewise aromatic ketones with substituents in the ortho positions resulted in no conversion (6k, 6m).
Table 1. Hydrosilylation of ketones utilizing complex 7 as catalyst.a

a Reaction conditions: 0.67 mmol ketone, 1.35 mmol Si(OMe)3H (2 equiv), 1 mol % catalyst, 3 cm3 toluene, 1 h, 25oC. Isolated yields are reported.
In sum, we prepared and characterized several new Cr(II) and Cr(III) NCN pincer complexes with an arene backbone connected to amine donors NEt2 and NiPr2via CH2-linkers. The new complexes were typically prepared in a one-pot synthesis by reacting the in situ lithiated ligand precursors N(C-Br)NCH2-Et (1) and N(C-Br)NCH2-iPr (2) with the Cr(III) and Cr(II) precursors [CrCl3(THF)3] and anhydrous CrX2 (X = Cl, Br), respectively. In the case of Cr(III), complex trans-[Cr(κ3NCN-NCNCH2-Et)(Cl)2(THF)] (3) was obtained. In the case of Cr(II), the reaction of lithiated 2 with a suspension of anhydrous CrCl2, complex [Cr(κ2NC-NCNCH2-iPr)2] (4) featuring two NCN ligands bound in κ2NC-fashion. If lithiated 2 is treated with a homogeneous solution of anhydrous CrX2 (X = Cl, Br) complexes [Cr(κ3NCN-NCNCH2-iPr)X] (5a,b) are obtained where the NCN ligand is coordinated in the typical meridional κ3NCN-mode. Treatment of 5a with 1 equiv of PhCH2MgCl and LiCH2SiMe3 afforded the alkyl complex [Cr(κ3NCN-NCNCH2-iPr)(CH2Ph)] (6) and [Cr(κ3NCN-NCNCH2-iPr)(CH2SiMe3)] (7), respectively. If a solution of lithiated 2 is treated with CrCl2 followed by addition of an excess of Na[HB(Et)3] the dimeric complex [Cr(κ2NC-NCNCH2-iPr)(µ2-H)]2 (8) is formed bearing two µ2-hydride ligands bridging the two Cr(II) centers. This complex displays a solution magnetic moment of µeff = 6.9(3) µB which is consistent with eight unpaired electrons as expected for a high-spin d4 configuration of the two metal centers and no metal-metal interaction. This finding was also confirmed by DFT calculations. Finally, the alkyl complex [Cr(κ3NCN-NCNCH2-iPr)(CH2SiMe3)] (7) turned out to be catalytically active for the hydrosilylation of aromatic and aliphatic ketones with Si(OMe)3H at room temperature with a catalyst loading of 1 mol%. X-ray structures of all complexes are presented.
All manipulations were performed under an inert atmosphere of argon by using Schlenk techniques or in a MBraun inert-gas glovebox. The solvents were purified according to standard procedures [29]. The deuterated solvents were purchased from Aldrich and dried over 3 Å molecular sieves. The ligands 1,1’-(2-bromo-1,3-phenylene)-bis-(N,N-diethylamine) (N(C-Br)NCH2-Et) (1) and 1,1’-(2-bromo-1,3-phenylene)-bis-(N,N-diisopropylamine) (N(C-Br)NCH2-iPr) (2) and anhydrous CrBr2 were synthesized according to literature [30–32]. All other materials are known compounds and were used as obtained from commercial suppliers. 1H, 13C{1H} and 19F{1H} NMR spectra were recorded on Bruker AVANCE-250, AVANCE-400, and AVANCE-600 spectrometers. 1H and 13C{1H} NMR spectra were referenced internally to residual protio-solvent and solvent resonances, respectively, and are reported relative to tetramethylsilane (δ = 0 ppm). 19F{1H} NMR spectra were referenced externally to CFCl3.
High resolution-accurate mass spectra were recorded on a hybrid Maxis Qq-aoTOF mass spectrometer (Bruker Daltonics, Bremen, Germany) fitted with an ESI-source. Measured accurate mass data of the [M]+ ions for confirming calculated elemental compositions were typically within 5 ppm accuracy. The mass calibration was done with a commercial mixture of perfluorinated trialkyl-triazines (ES Tuning Mix, Agilent Technologies, Santa Clara, CA, USA).
Electron Paramagnetic Resonance (EPR) spectra were recorded on an X-band Bruker Elexsys-II E500 CW-EPR spectrometer (Bruker Biospin GmbH, Rheinstetten, Germany) equipped with a high sensitivity cavity (SHQE1119) at 100 ± 1 K. The instrument parameters were set as follows: microwave frequency, 9.43 GHz; modulation frequency, 100 kHz, and microwave power, 15.9 mW. The spectra were analyzed using Xepr software and the Anisotropic SpinFit simulation program (both Bruker Biospin GmbH).
Synthesis of [2,3-bis[(diethylamino)methyl]phenyl- C,N,N′](dichloro) (tetrahydrofuran)chromium(III), [Cr(κ3NCN-NCNCH2-Et)(Cl)2(THF)] (3)
A solution of (N(C-Br)NCH2-Et) (1) (400 mg, 1.23 mmol) in THF (13 cm3) was cooled to -90°C. After stirring for 10 minutes, n-BuLi (1.52 cm3, 1.6 M in hexanes, 2.46 mmol) was added dropwise via a syringe and the light-yellow solution was stirred at -90°C for 2.5 h. After warming up to -30°C, [CrCl3(THF)3] (460 mg, 1.23 mmol) was added portion-wise under stirring resulting in the formation of a dark green solution. The solution was allowed to warm to room temperature and was stirred for 12 h. After removal of the solvent under reduced pressure, the residue dissolved in toluene (7 cm3) and filtered through celite. After removal of the toluene, the residue was washed three times with n-pentane (3x10 cm3). Drying under reduced pressure yielded a light-purple solid. Yield: 340 mg, 63%). Dark green crystals suitable for X-ray crystallography were obtained by slow diffusion of n-pentane into a saturated THF solution of 3. µeff = 3.7(2)µB (Evans Method, THF), HR-MS (ESI+, THF) m/z calcd for C16H27Cl2CrN2 [M-THF + H]+ 370.1029, found 370.1031.
Synthesis of Bis-[2,3-bis[(di iso propylamino)methyl]phenyl- C,N,N′]chromium(II), [Cr(κ2NC-NCNCH2-iPr)2] (4)
A solution of (N(C-Br)NCH2-iPr) (2) (100 mg, 0.26 mmol) in THF (7 cm3) was cooled to -90° C. After stirring at that temperature for 10 min, n-BuLi (0.18 cm3, 1.6 M, 0.30 mmol) was added dropwise via a syringe. A suspension of anhydrous CrCl2 (38 mg, 0.30 mmol) in THF (4 cm3) was added in a dropwise fashion at -20°C. After the addition was completed, all volatiles were removed under reduced pressure and the residue was extracted with n-pentane (6 cm3) and filtered through a syringe filter (PTFE, 0.2 µm). The solvent was removed under vacuum yielding a dark blue solid. Yield: 45 mg (37%). Blue crystals suitable for X-ray crystallography were obtained by cooling a saturated n-pentane solution of 4 to -30° C. µeff = 4.7(3) µB (Evans Method, benzene). HR-MS spectra could not be obtained due to the highly air-sensitive nature of the compound.
Synthesis of [2,3-bis[(di iso propylamino)methyl]phenyl- C,N,N′](chloro) chromium(II), [Cr(κ3NCN-NCNCH2-iPr)Cl] (5a)
Anhydrous CrCl2 (38 mg, 0.30 mmol) was suspended in THF (15 cm3). The suspension was exposed to ultrasonic irradiation for 1 h to form a light-blue clear solution. The ligand (N(C-Br)NCH2-iPr) (2) (100 mg, 0.26 mmol) was dissolved in THF (5 cm3) and subsequently cooled to -90° C. After stirring at that temperature for 10 minutes, n-BuLi (0.18 cm3, 1.6M, 0.30 mmol) was added dropwise via a syringe. The orange solution was stirred for 30 min before it was allowed to warm to 0° C while stirring another hour. The lithiated intermediate was added in a dropwise fashion to the THF-solution of CrCl2via a syringe. The solution was allowed to stir for 15 min at room temperature before all volatiles were evaporated under vacuum. The residue was extracted into n-pentane (7 cm3) and filtered through a syringe filter (PTFE, 0.2 µm). The volume of the filtrate was reduced to about 1.5 cm3 and stored at -30° C to afford 5 as dark purple crystalline plates suitable for X-ray diffraction analysis. Yield: 75 mg (73%). µeff = 4.8(2) µB (Evans Method, benzene). HR-MS spectra could not be achieved due to the highly air-sensitive nature of the compound.
Synthesis of [2,3-bis[(di iso propylamino)methyl]phenyl- C,N,N′](bromo) chromium(II), [Cr(κ3NCN-NCNCH2-iPr)Br (5b)
Complex 5b was prepared analogously to complex 5a utilizing anhydrous CrBr2 (58 mg, 0.30 mmol) and N(C-Br)NCH2-iPr (2) (100 mg, 0.26 mmol) as starting materials. Yield: 85.2 mg (75%). µeff = 4.7(2) µB (Evans Method, benzene). HR-MS spectra could not be obtained due to the highly air-sensitive nature of the compound.
Synthesis of [2,3-bis[(di iso propylamino)methyl]phenyl- C,N,N′](phenyl)(methyl)) chromium(II), [Cr(κ3NCN-NCNCH2-iPr)(CH2Ph)](6)
Method A: A suspension of anhydrous CrCl2 (38 mg, 0.30 mmol) in THF (15 cm3) was placed in an ultrasonic bath for 1 h whereupon the suspension turned into light blue solution. (N(C-Br)NCH2-iPr) (2) (100 mg, 0.26 mmol) was dissolved in THF (5 cm3) and subsequently cooled to -90° C. After stirring for 10 min, n-BuLi (0.18 cm3, 1.6M, 0.30 mmol) was added dropwise and the solution was stirred for additional 30 min. The solution was allowed to warm to 0° C and was stirred for 1 h. The lithiated intermediate was then added in a dropwise fashion to THF solution of CrCl2 and a dark colored solution was formed immediately. The solution was stirred for 15 min at room temperature and all volatiles were then evaporated under vacuum. The residue was dissolved in benzene and the solution was filtered through a syringe filter (PTFE, 0.2 µm). A solution of PhCH2MgCl (0.1 cm3, 1M in THF) was then added and the suspension was stirred for 1.5 h. All volatiles were removed under reduced pressure. The remaining residue was extracted with n-pentane (3 cm3) and filtered through syringe filter (PTFE, 0.2 µm). Evaporation of the solvent afforded 6 as a dark brown solid. Cooling of a saturated n-pentane solution of 6 to -30° C yielded crystals suitable for X-Ray diffraction. Yield: 81 mg (72%). µeff = 4.9 (1) µB (Evans Method, benzene).
Method B. Complex 6 was also obtained by reacting isolated 5a (50 mg, 0.12 mmol) in benzene (2 cm3) with PhCH2MgCl (0.14 cm3, 1 M in THF) and stirring for 1.5 h at room temperature. After evaporating the solvent under reduced pressure, redissolving the residue in n-pentane (2 cm3) and subsequent filtration (syringe filter PTFE, 0.2 µm) and removal of the solvent, 6 could be isolated as a dark brown solid. Yield: 41 mg (72%). HR-MS spectra could not be achieved due to the highly air-sensitive nature of the compound.
Synthesis of [2,3-bis[(di iso propylamino)methyl]phenyl- C,N,N′](trimethylsilane) (methyl)chromium(II), [Cr(κ3NCN-NCNCH2-iPr)(CH2SiMe3)] (7)
Method A. Complex 7 was prepared in analogous fashion to 6 utilizing LiCH2TMS (0.28 cm3, 0.28 mmol, 1 M in n-pentane). Complex 7 was obtained as dark brown solid. Yield: 76 mg (68%). µeff = 4.8 (1)µB (Evans Method, benzene).
Method B: A solution of Complex 5a (50 mg, 0.12 mmol) in benzene (2 cm3) was treated with LiCH2TMS (0.13 cm3, 0.28 mmol, 1 M in n-pentane) and stirring for 1.5 h at room temperature. After evaporation of the solvent, redissolving the residue in n-pentane (2 cm3) and subsequent filtration (syringe filter PTFE, 0.2 µm) and removal of the solvent, 7 could be isolated as a dark brown solid. Yield: 39 mg (69%). HR-MS spectra could not be obtained due to the highly air-sensitive nature of the compound.
Synthesis of Bis-[2,3-bis[(di iso propylamino)methyl]phenyl- C,N,N′])(µ2-hydrido) chromium(II)], [Cr(κ2NC-NCNCH2-iPr)(µ2-H)]2 (8)
A suspension of anhydrous CrCl2 (38 mg, 0.30 mmol) in THF (15 cm3) was placed in an ultrasonic bath for 1 h, whereupon a light blue solution was obtained. The ligand precursor N(C-Br)NCH2-iPr (2) (100 mg, 0.26 mmol) was dissolved in THF (5 cm3) and cooled to -90° C. After stirring for 10 min, n-BuLi (0.18 cm3, 1.6 M, 0.30 mmol) was added dropwise via a syringe and the orange solution was further stirred for 30 min and then allowed to warm to 0° C and stirred for an additional 1 h. The solution containing lithiated N(C-Br)NCH2-iPr (2) was added dropwise to the THF-solution of anhydrous CrCl2 whereupon the solution became dark purple. The solution was allowed to stir for 15 min at room temperature before all volatiles were evaporated under vacuum. The residue was dissolved in benzene and filtered through a syringe filter (PTFE, 0.2 µm) and volatiles were evaporated under reduced pressure. The residue was dissolved in precooled THF (-30°C, 3 cm3) and treated with NaHBEt3 (0.91 cm3, 0.91 mmol, 1M in THF) and stirred for 30 min. After removal of the solvent, the remaining residue was redissolved in n-pentane (10 cm3), filtered through a syringe filter (PTFE, 0.2 µm) and the volume of the solution was reduced to approximately 1 cm3. Storage of the light brown solution at -30° C for 12 h led to the formation of bright orange crystals. Yield: 64 mg (35%). µeff = 6.9 (3) µB (Evans Method, benzene-d6). HR-MS (ESI+, THF) m/z calcd for C40H72Cr2N4 [M-C20H36CrN2]+ 356.2279, found 356.2278.
General Procedure for the Hydrosilylation of Ketones
To a solution of the substrate (0.67 mmol, 1 equiv.) and HSi(OMe)3 (174 mm3, 1.35 mmol, 2 equiv) in toluene (3 cm3) complex 6 was added (3 mg, 6.7 µmol, 1 mol%) causing an immediate color change. The mixture was stirred at room temperature for 1 h and then a saturated methanolic K2CO3 solution (3 cm3) was added to the mixture. After stirring for 5 h volatiles were evaporated under reduced pressure. The residue was dissolved in CH2Cl2 (6 cm3) and filtered through a short plug of silica. After evaporation of the solvent the reaction products were analyzed by the means of 1H, 13C {1H} and 19F{1H} NMR spectroscopy.
X-ray Structure Determination
X-ray diffraction data of 3, 4, 5a (CCDC 2285377–2285379) and 8 (CCDC 2285381) were collected at T = 100 K in a dry stream of nitrogen on a Bruker Kappa APEX II diffractometer system using graphite-monochromatized Mo-Kα radiation (λ = 0.71073 Å) and fine sliced φ- and ω-scans. Data were reduced to intensity values with SAINT and an absorption correction was applied with the multi-scan approach implemented in SADABS [33]. Data of 6 (CCDC 2285380) were collected at T = 100 K on a Rigaku XtaLAB Synergy, Dualflex diffractometer system equipped with a HyPix hybrid photon counting detector using Cu-Kα radiation (λ = 1.54184 Å). Data were reduced and an absorption correction applied using the multi-scan approach with the CrysAlisPro software [34]. The structures were solved by the dual-space approach implemented in SHELXT [35] and refined against F2 with SHELXL [36]. Non-hydrogen atoms were refined with anisotropic displacement parameters. H atoms attached to C were placed in calculated positions and thereafter refined as riding on the parent atoms. The positions of the hydride Hs in 8 were freely refined. The halogenide ligands in 3 and 5a were modelled as occupationally disordered Cl/Br sites with the total occupation of each sit e constrained to 1. Molecular graphics were generated with the program MERCURY [37].
Computational Details
The computational results presented have been achieved in part using the Vienna Scientific Cluster (VSC). Calculations were performed using the GAUSSIAN 09 software package [38] and the OPBE [39–44] functional without symmetry constraints, the Stuttgart/Dresden ECP (SDD) basis set to describe the electrons of the chromium atom and a standard 6-31G** basis for all other atoms as already previously described [45]. Population Analysis (NPA) [46] and the resulting Wiberg indices [28] were used to study the electronic structure and bonding of the optimized species. The NPA analysis was performed with the NBO 5.0 program [47]
Electronic supplementary material. The online version of this article (https://doi.org/xxxxxxxxxxx) contains supplementary material, which is available to authorized users.
Acknowledgements
Financial support by the Austrian Science Fund (FWF) is gratefully acknowledged (Project P 32570-N). The X-Ray center of the Vienna University of Technology is acknowledged for financial support and for providing access to the single-crystal diffractometer. We thank Christian Göb of Rigaku Europe SE for performing the single crystal diffraction experiment of complex 6.
- Gossage RA, van de Kuil LA, van Koten G (1998) ) Acc Chem Res 31:423
- Albrecht M, van Koten G (2001) Angew Chem Int Ed 40:3750
- van der Boom ME, Milstein D (2003) Chem Rev 103:1759
- Liang LC (2006) Coord Chem Rev 250:1152
- Morales-Morales D, Jensen CM (2007) The Chemistry of Pincer Compounds. Elsevier
- Nishiyama H (2007) Chem Soc Rev 36:1133
- Benito-Garagorri D, Kirchner K (2008) Acc Chem Res 41:201
- Choi J, MacArthur AHR, Brookhart M, Goldman AS (2011) Chem Rev 111:1761
- Selander N, Szabo KJ (2011) Chem Rev 111:2048
- Schneider S, Meiners J, Askevold B (2012) Eur J Inorg Chem 2012:412
- van Koten G, Milstein D (2013) Organometallic Pincer Chemistry. Springer, Berlin Heidelberg
- Murugesan S, Kirchner K (2016) Dalton Trans 45:416
- Grove DM, van Koten G, Ubbels HJC, Zoet R (1984) Organometallics 3:1003
- Slagt MQ, van Zwieten DAP, Moerkerk AJCM, Klein Gebbink RJM, van Koten G (2004) Coord Che Rev 248:2275
- Back S, Gossage RA, Lang H, van Koten G (2000) Eur J Inorg Chem 1457
- Rodriguez G, Lutz M, Spek AL, van Koten G (2002) Chem Eur J 8:45
- van der Zeijden AAH, van Koten G, Luijk R, Vrieze K, Slob C, Krabbendam H, Spek AL (1988) ) Inorg Chem 27:1014
- Schimmelpfennig U, Simmering RZ, Schleinitz KD, Stößer R, Wenschuh E (1993) Z Anorg Allg Chem 619:1931
- Adams JJ, Arulsamy N, Roddick DM (2012) Organometallics 31:1439
- Pecak J, Eder W, Tomsu G, Pignitter M, Kirchner K (2021) Eur J Inorg Chem 41:4280
- Himmelbauer D, Mastalir M, Stöger B, Veiros LF, Kirchner K (2018) ) Organometallics 37:3631
- Himmelbauer D, Stöger B, Pignitter M, Veiros LF, Kirchner K (2019) Organometallics 38:4669
- Eder W, Stöger B, Kirchner K (2019) Monatsh Chem 150:1235
- Ott JC, Isak D, Melder JJ, Wadepohl H, Gade LH (2020) Inorg Chem59:14526
- Schiwek CH, Vasilenko V, Wadepohl H, Gade LH (2018) Chem Commun 54:9139
- Liu Z, Gao W, Liu X, Luo X, Cui D, Mu Y (2011) Organometallics 30:752
- Addison AW, Rao TN, Reedijk J, van Rijn J, Verschoor GC (1984) J Chem Soc Dalton Trans 1349
- Wiberg KB (1968) Tetrahedron 24:1083. Wiberg indices are electronic parameters related with the electron density in between two atoms, which scale as bond strength indicators. They can be obtained from a Natural Population Analysis
- Perrin DD, Armarego WLF (1988) Purification of Laboratory Chemicals, 3rd edn. Pergamon, New York
- van Beek JAM, van Koten G, Dekker GPCM, Wissing E, Zoutberg MC, Stam CH (1990) J Organomet Chem 394:659
- Bibal C, Mazières S, Gornitzka H, Couret C (2002) Polyhedron 21:2827
- Kurogi T, Irifune K, Takai K (2021) Chem Sci 12:14281
- Bruker computer programs (2020) APEX3, SAINT, SADABS. Bruker AXS Inc., Madison, WI)
- Rigaku OD (2022) CrysAlisPro software system version 1.171.42.69a. Rigaku Corporation, Wrocław, Poland
- Sheldrick GM (2015) Acta Crystallogr A 71:3
- Sheldrick GM (2015) Acta Crystallogr C 71:3
- Macrae CF, Edgington PR, McCabe P, Pidcock E, Shields GP, Taylor R, Towler M, van de Streek J (2006) J Appl Cryst 39:453
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman G Jr, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09, revision A.02. Gaussian Inc., Wallingford.
- Handy NC, Cohen AJ (2001) Mol Phys 99:403
- Hoe WM, Cohen A, Handy NC (2001) Chem Phys Lett 341:319
- Perdew JP, Burke K, Ernzerhof M (1997) Phys Rev Lett 78:1396
- Perdew JP, Burke K, Ernzerhof M (1996) Phys Rev Lett 77:3865
- Swart M (2008) J Chem Theory Comput 4:2057
- Conradie J, Ghosh A (2007) J Chem Theory Comput 3:689
- Pecak J, Stöger B, Veiros LF, Ferreira LP, Pignitter M, Linert W, Kirchner K (2019) Inorg Chem 58:4641
- Reed AE, Weinstock RB, Weinhold F (1985) J Chem Phys 83:735
- NBO 5.0, Glendening ED, Badenhoop JK, Reed AE, Carpenter JE, Bohmann JA, Morales CM, Weinhold F (2001) Theoretical Chemistry Institute, University of Wisconsin, Madison
Schemes 1 and 5 are available in the Supplementary Files section
- GA.png
Graphical Abstract
- Scheme1.png
Scheme 1. Examples of chromium complexes featuring anionic PCP, PNP, NNN and NCN ligands
- Scheme2.png
Scheme 2. Synthesis of Complex 3
- Scheme3.png
Scheme 3. Synthesis of complexes 4 and 5
- Scheme4.png
Scheme 4. Synthesis of complexes 6 and 7
- Scheme5.png
Scheme 5. Synthesis of complex 8
- SupportingInformation.pdf
- checkcif.pdf
- all.cif
- CrNCNDimer8.xyz
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}