HO‑PBA dimer
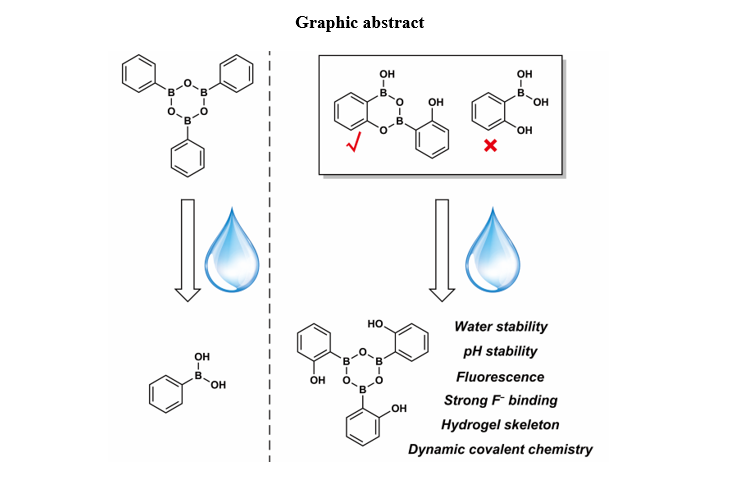
HO‑PBA is a useful organic molecule that is widely employed in organic reactions and catalysis17,23,24. However, its single-crystal structure, which enables the visualization of intermolecular interactions and provides insights into stereochemistry and reaction selectivity, is currently unavailable. Yet, these pieces of information are crucial for the explanation of the reaction and catalysis mechanisms. Herein, we successfully obtained the single crystal of HO‑PBA through slow evaporation of its solution. Surprisingly, the single-crystal structure of HO‑PBA challenges the notion in the literature that HO‑PBA exists as a monomer16,17. Instead, it clearly displays that HO‑PBA exists as a unique dimer through the formation of two B–O covalent bonds (Fig. 2a), which was also confirmed by the 1H and 13C NMR spectra (Fig. 3b,c). Additionally, this dimeric structure was observed in other derivatives of HO-PBA, such as 2-hydroxy-5-methylphenylboronic acid (CH3-HO‑PBA) with the electron-donating group –CH3 (Supplementary Fig. 1a) and 2-hydroxy-4-(trifluoromethyl)phenylboronic acid (CF3-HO‑PBA) with the electron-withdrawing group –CF3 (Supplementary Fig. 2a). This suggests that the formation of dimeric structures might be a common characteristic of HO‑PBA and its derivatives.
DCBs in the HO‑PBA dimeric structure
Further investigations unveiled an intriguing aspect regarding the dimeric structure of HO‑PBA, wherein the two B–O covalent bonds are DCBs that exhibit reversible breakage and formation properties. This was evidenced by the rapid exchange between different HO‑PBA dimeric structures at room temperature. To observe this exchange reaction, HO‑PBA dimers and CH3-HO‑PBA dimers were co-dissolved in a dry THF solution at room temperature. After evaporation of THF, the resulting powder was characterized using matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) MS. The mass spectrum shows two peaks at m/z 239.18 and 267.22 that correspond to HO‑PBA dimer and CH3-HO‑PBA dimer, respectively (D0 and D2), and a third peak at m/z 253.20, attributed to their exchange product (D1) (Fig. 2b,c). This indicated that an exchange reaction occurred between HO‑PBA dimer and CH3-HO‑PBA dimer, resulting in a mixture of homo- and hetero-substituted HO‑PBA dimeric structure under equilibrium. Similar exchange reactions were also observed between HO‑PBA dimer and CF3-HO‑PBA dimer (Supplementary Fig. 3), as well as between CF3-HO‑PBA dimer and CH3-HO‑PBA dimer (Supplementary Fig. 4). DCBs provide a powerful avenue to synthesize complex molecular architectures (e.g., cages, macrocycles and interlocked molecules) 25–27, and materials with advanced properties (e.g., COFs, self-healing materials, and vitrimers)28,29. The highly dynamic nature of B–O bonds in the HO‑PBA dimeric structure at room temperature gives rise to its tremendous potential in these aspects.
AIEE property of HO‑PBA dimer
In the single-crystal structure, HO‑PBA dimers aggregate into an ordered packing by means of intermolecular O–H…O and C–H…O hydrogen bonds (Fig. 2d,e, Supplementary Fig. 5 and Supplementary Tables 1–3). Correspondingly, the scanning electron microscopy (SEM) image of the HO‑PBA dimer sample, which was prepared by dropping the HO‑PBA solution onto a clean silicon wafer, displays that HO‑PBA dimers self-assemble into flower-shaped structures (Fig. 2f). Moreover, the solid HO‑PBA dimer sample (Aladdin Reagent, Shanghai, Product No. H101964) has a cuboid shape and exhibits sharp reflection peaks in its X-ray diffraction (XRD) pattern (Fig. 2g), illustrating its crystalline nature. The highly ordered molecular packing and crystalline structure suggest that the intramolecular motions of HO‑PBA dimers might be restricted in the aggregate, leading us to speculate that HO‑PBA dimers might exhibit AIEE, a well-known photophysical phenomenon characterized by a stronger emission of molecular aggregates than that of individual molecules30,31. As shown in Fig. 2h, the solid HO‑PBA dimer sample emits strong fluorescence (red line) and possesses an absolute quantum yield (ΦF) of 73.0%, significantly higher than that of HO‑PBA dimer in THF (absolute ΦF: 9.7%). These fluorescent results confirmed the AIEE property of HO‑PBA dimers. Likewise, CH3-HO‑PBA and CF3-HO‑PBA dimers also exhibit AIEE activities (Supplementary Figs. 1, 2 and 6–8 and Supplementary Tables 4–9). Typically, conventional aromatic AIEE molecules possess highly twisted spatial configurations and bulky molecular sizes, such as hexaphenylsilole, tetraphenylethene, and their derivatives18,19. However, despite its simple molecular structure, HO‑PBA dimers display AIEE activity with a remarkably high absolute ΦF, introducing fresh insights into the AIEE realm.
Discovery of a water-stable boroxine structure
The AIEE phenomenon is usually exhibited by a gradually enhanced fluorescent emission when the fraction of water (poor solvent) to organic solvent (good solvent, such as THF and methanol) is increased32. However, the HO‑PBA dimer’s characteristic fluorescent peak at 316 nm in the THF–water solution shows a slow upward trend when the volume fraction of water increases from 0–30%, after which it gradually decreases (Fig. 3a). Meanwhile, a new fluorescent peak at 368 nm appears, and its relative intensity (F/Fmax) gradually increases from 0.1 to 1 with the increase of the volume fraction of water (Fig. 3a, the absolute ΦF of HO‑PBA dimer in water is 19.4%). Similar fluorescent variations of HO‑PBA dimer were observed in methanol–water solutions with different water fractions (Supplementary Fig. 9), indicating that the fluorescent variations of HO‑PBA dimer are induced by water and independent of the organic solvent employed.
To investigate changes in the HO-PBA dimer solution resulting from the addition of water, NMR characterizations were conducted. The 1H (Fig. 3b) and 13C (Fig. 3c) NMR spectra of HO-PBA dimer in THF − d8 display all the signals of HO-PBA dimer, except for the signals of the two C atoms bonded to B atoms (Fig. 3c, marked with 1 and 7 in the molecular structure of HO-PBA dimer) due to the quadrupolar relaxation mechanism of 11B nucleus33. However, upon addition of water into the THF − d8 solution of HO-PBA dimer, 1H (Fig. 3d) and 13C (Fig. 3e) NMR signals are reduced to four and five sets, respectively, which are consistent with the numbers of H and C present in HO‑PBA monomer (The signal of C atom bonded to B atom is too weak to be detected.). Thus, we hypothesized that HO‑PBA dimer hydrolyzed into HO‑PBA monomer in the present of water. To test this hypothesis, 10-hydroxybenzo[h]quinolone (HBQ), an effective reagent for detecting boronic acid groups, was employed34. Excited HBQ undergoes an excited-state intramolecular proton transfer (ESIPT) process with the maximum emission at 597 nm. Binding with boronic acid groups would interrupt the ESIPT of HBQ, resulting in a shift of the maximum emission to 504 nm35. After mixing HBQ with excess HO‑PBA dimers in the solution, the emission peak at 504 nm did not appear (Fig. 3f), which implies no boronic acid groups are present in the water–THF solution of HO‑PBA dimer. Therefore, our hypothesis about the transformation of HO-PBA dimer into monomer in the presence of water is incorrect.
The changes in the HO-PBA dimer’s solution subsequent to the addition of water were further examined using electrospray ionization quadrupole time-of-flight (ESI-Q-TOF) MS. In Fig. 4a, two strong peaks at m/z 359.13 and 373.14 captured our attention. The m/z 359.13 corresponds to the molecular weight of the deprotonated HO‑PBA trimer ([T − H]−), while the m/z 373.14 corresponds to the molecular weight of the deprotonated product [T + CH2 − H]− from the HO‑PBA trimer’s etherification with the solvent methanol during the MS measurement. This indicates that HO‑PBA dimer might transform into HO‑PBA trimer in the presence of water. The 1H and 13C NMR spectra of HO‑PBA trimer display four and five sets of signals (Fig. 3d,e), respectively, suggesting that HO‑PBA trimer has a symmetric structure. Based on these findings, we boldly speculated that HO‑PBA trimer was a molecule with a boroxine structure, which was stable in aqueous media, as depicted in Fig. 4a.
Characterization of the molecular structure of HO‑PBA trimer
Firstly, the presence of phenolic hydroxyl groups in the molecular structure of HO-PBA trimer was examined by the typical reaction between phenolic hydroxyl groups with Br2 or Fe3+. Upon mixing the HO‑PBA trimer solution with bromine water, a white precipitate was formed (Fig. 4b1). Besides, the addition of freshly prepared ferric chloride solution turned the initially colorless HO‑PBA trimer solution into purple (Fig. 4b2). These results prove that the phenolic hydroxyl groups are preserved in the molecular structure of HO‑PBA trimer.
Secondly, UV–Raman spectrometry was employed to investigate whether a boroxine ring is present in the molecular structure of HO‑PBA trimer. For comparison, the UV–Raman spectra of TPB and PBA were also measured. The solid TPB and PBA sample share four UV–Raman peaks at 995, 799, 760, and 661 cm− 1 (Fig. 4c, black and red line). The peak at 995 cm− 1 is attributed to the stretching vibration of B–O groups and the bending vibration of B–OH groups36,37. Due to the ease of dehydration, PBA readily transforms into TPB, resulting in the coexistence of PBA and TPB in the solid PBA sample. Thus, the remaining three peaks could be reasonably assigned to the characteristic vibration of the boroxine ring in TPB. When exposed to water, TPB hydrolyzed into PBA, and the three peaks disappeared (Fig. 4c, green and blue line). This result further supported the assignment of the three peaks to the characteristic vibration of the boroxine ring.
With the characteristic peaks of boroxine ring being confirmed, we focused our attention on the UV–Raman spectrum of the solid HO‑PBA dimer sample. Due to the asymmetry of its molecular structure, the solid HO‑PBA dimer sample exhibits weak UV–Raman peaks (Fig. 4d, black line). Conversely, when HO‑PBA dimer was dissolved in an acetonitrile (ACN)–water solution, four distinct UV–Raman peaks appeared at 1032, 835, 705, and 579 cm− 1, respectively (Fig. 4d, red line), besides the peak at 922 cm− 1 assigned to the C-C stretching of ACN (Fig. 4c, cyan line). Since HO‑PBA dimer transforms into trimer in the presence of water, these four peaks belong to HO‑PBA trimer. The discrepancies in peak positions between HO-PBA trimer and the solid TPB sample in the UV-Raman spectra could be attributed to the influence of the phenolic hydroxyl substituents on the skeletal vibration of HO-PBA trimer. The above results confirmed the presence of a boroxine ring within the molecular structure of HO‑PBA trimer.
Lastly, density functional theory (DFT) calculations further supported the HO‑PBA trimer’s structure. As displayed in Fig. 4e, the central boroxine skeleton adopts a hexagonal shape that is nearly flat and regular with bond angles close to 120°. The arrangement of the three phenyl rings that are connected to B atoms is found to be non-planar relative to the boroxine ring. Furthermore, intramolecular hydrogen bonding is observed between the boroxine ring and the phenolic hydroxyl groups. Based on the optimized geometry, the NMR and fluorescent spectrum of HO‑PBA trimer in water were calculated. The calculated 1H and 13C NMR spectra of HO‑PBA trimer are in agreement with the experimental data (Supplementary Fig. 10). The calculated fluorescent spectrum displays two bands (P1 and P2) located at 328 and 364 nm, respectively, which is also consistent with the experimental data (Fig. 4f). The P1 fluorescence is characteristic of local-excited emission, as determined by the fact that the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of P1 are mainly localized on the same phenyl fragment (Fig. 4g). The HOMO and LUMO of P2 are distributed on the different fragments of HO‑PBA trimer, indicating that the P2 emission originates from the charge-transfer state (Fig. 4g)38,39.
It is worth noting that the transformation from HO‑PBA dimer to trimer occurs rapidly at room temperature. Following addition of water to the THF–d8 solution of HO‑PBA dimer, a 1H NMR measurement was performed immediately. The resulting 1H NMR spectrum clearly indicated that HO‑PBA dimer had completely transformed into trimer at room temperature. The same changes were also observed for CH3-HO-PBA and CF3-HO-PBA dimers (Supplementary Figs. 11–16). Moreover, fluorescence measurements indicate that the boroxine structure remains stable over a wide pH range, specifically, 2.4༜pH༜9 for HO‑PBA trimer (Fig. 4h), and 2.4༜pH༜10 for CH3-HO‑PBA trimer (Supplementary Fig. 11h). The remarkable stability of the boroxine structure in water and its wide pH tolerance range make it suitable for various applications where water is present.
Role of water in the formation of the boroxine structure
The transformation of HO‑PBA dimer to trimer in water prompts an interesting question: does water participate in this process? It is impossible to determine water’s involvement solely from the reaction equation because HO‑PBA dimer and trimer share the same empirical formula. To address this question, the isotope tracing method was employed by dissolving HO‑PBA dimer in a methanol–H218O (5:1, v/v) solution and subsequently measuring ESI-Q-TOF MS. In the methanol–H2O solution, HO‑PBA trimer exhibits two peaks at m/z 359.13 and 373.14 (Fig. 4a), whereas in the methanol–H218O solution, the peaks shift to m/z 365.11 and 377.13 (Fig. 4i). The m/z 365.11 corresponds to the molecular weight of the deprotonated HO‑PBA trimer with three 18O ([T\318O−H]−), while the m/z 377.13 corresponds to the molecular weight of the deprotonated product with two 18O ( [T\218O+CH2 − H]−), resulting from the etherification of HO‑PBA trimer and the solvent methanol during the MS measurement. This result implied that H218O participated in the transformation process of HO‑PBA dimer to trimer.
Upon removal of the solvent of a H2O–THF (2:1, v/v) solution of HO‑PBA trimer, the residue was identified as HO‑PBA dimer by its 1H NMR spectrum in THF − d8 (Supplementary Fig. 17). This result suggests that the presence of water is essential for the existence of HO‑PBA trimers, and they spontaneously transform into dimers in the absence of water (Fig. 1b). Therefore, water not only participates in the transformation process of HO‑PBA dimer to trimer, but also provides a necessary environment for the stable existence of HO‑PBA trimer. It remains challenging to differentiate the roles of water in the dimer-to-trimer reaction and its contribution to the stabilization of HO‑PBA trimer. To tackle this challenge, we are currently exploring these mechanisms through detailed reaction kinetics studies and accurate theoretical calculations, hoping to unravel the answers in the future.
Water-compatible DCBs
DCBs have been firmly integrated into diverse research fields, such as organic synthesis, material science, and biomedicine. Nonetheless, most of DCBs are water-incompatible, which restricts their applicability to certain specific fields 28,40. Herein, we reveal that B–O bonds in HO‑PBA trimer are water-compatible DCBs, providing a significant member to the DCBs family.
The dynamic nature of B–O bonds in the water-stable boroxine structure was illustrated by the rapid exchanges between various HO‑PBA trimeric structures. To carry out the exchange reaction, HO‑PBA dimers and CF3-HO‑PBA dimers were co-dissolved in a methanol–water solution at room temperature, and the solution was then analyzed by ESI-Q-TOF MS. In addition to the previously identified peaks of HO‑PBA trimer ([T − H]− at m/z 359.13 and [T + CH2 − H]− at m/z 373.14) and CF3-HO‑PBA trimer ([T3 − H]− at m/z 563.11 and [T3 + CH2 − H]− at m/z 577.13), peaks related to their exchange products ([T1 − H]− at m/z 427.12, [T1 + CH2 − H]− at m/z 441.14, [T2 − H]− at m/z 495.11, and [T2 + CH2 − H]− at m/z 509.13) were also present in the mass spectrum (Fig. 5). This suggested an exchange reaction occurred between HO‑PBA trimer and CF3-HO‑PBA trimer, resulting in a mixture of homo- and hetero-substituted HO‑PBA trimeric structure under equilibrium. Similar exchange reactions were also observed between HO‑PBA trimer and CH3-HO‑PBA trimer (Supplementary Fig. 18), as well as between CF3-HO‑PBA trimer and CH3-HO‑PBA trimer (Supplementary Fig. 19). These results verified the highly dynamic nature of B–O bonds in the boroxine structure in aqueous conditions at room temperature. The water-incompatible B–O bonds in the conventional boroxine structure are also dynamic, however, to carry out the exchange reactions, their toluene solutions need to be heated at 60○C for 8 hours41. By comparison, the water-compatible B–O bonds in the water-stable boroxine structure can rapidly exchange at room temperature, which suggests the flexibility of these structures in various applications.
Recognition of F− by the boroxine structure
Boroxine rings have been identified as effective anion receptors for F− in lithium ion batteries owing to their unique ring structures and electron-deficient properties 4,42. However, the applicability of boroxine rings as anion receptors in aqueous media is limited due to their hydrolytic instability43. Fortunately, the discovery of the water-stable boroxine structure provides a feasible solution to this challenge.
As shown in Fig. 6a, the 1H NMR spectrum of HO‑PBA trimer contains four sets of peaks, whereas more than ten sets of peaks appear after the addition of F−. In contrast, the addition of chloride ions (Cl−), bromide ions (Br−), or iodine ions (I−) did not cause any alterations to the 1H NMR spectrum of HO‑PBA trimer. These results confirmed that HO‑PBA trimer could selectively recognize F– in aqueous media.
The binding affinity of HO‑PBA trimer to F− in aqueous media was evaluated by comparing it with the commonly used F− recognition molecule, PBA20–22. Initially, a NaF suspension was prepared in a H2O–dimethylsulfoxide (DMSO) (1:6, v/v) mixture (Fig. 6b, left panel). In the 19F NMR spectrum of the NaF suspension (Fig. 6c, black line), an extremely weak signal at − 100.0 ppm attributed to free F− is observed, indicating that only a small amount of F− has dissolved in the suspension. Upon adding one molar equivalent of PBA, the NaF/PBA remained as a suspension (Fig. 6b, middle panel). In the 19F NMR spectrum of the NaF/PBA suspension (Fig. 6c, red line), a new yet extremely weak signal at − 135.2 ppm attributed to the PBA–F− complex appears, indicating that only a small amount of PBA–F− complex was formed in the NaF/PBA suspension. Due to the low content of the PBA–F− complex, the 11B NMR spectrum of the NaF/PBA suspension (Fig. 6d, red line) shows no change compared with that of the PBA solution (Fig. 6d, black line, Top). In sharp contrast, the NaF suspension became clear after adding one molar equivalent of HO‑PBA trimer (Fig. 6b, right panel). In the 19F NMR spectrum of the NaF/HO‑PBA trimer solution (Fig. 6c, blue line), the signal of free F− disappears, and multiple strong signals resulting from the equilibrations of the HO‑PBA trimer–F− complex and HO‑PBA trimer (Supplementary Fig. 20)44 appear in the − 125 to − 136 ppm range. Correspondingly, in the 11B NMR spectrum of the NaF/HO‑PBA trimer solution (Fig. 6d, blue line), strong sp3–11B signals of HO‑PBA trimer–F− complex (3.1 and 1.2 ppm) are observed, apart from the sp2–11B signal (28.7 ppm) 44,45. Similar results were obtained for CH3-HO‑PBA trimer (Supplementary Fig. 21) and CF3-HO‑PBA trimer (Supplementary Fig. 22). These findings indicate that the boroxine structure exhibits significantly stronger binding affinity to F− than PBA does, illustrating its good potential in F− detection and separation.
Boroxine-based dynamic hydrogel
Hydrogels have gained significant attention in recent years because of their potential applications in tissue engineering, drug delivery, biosensing, and other areas46,47. Herein, we designed a boroxine-based hydrogel to demonstrate the exceptional performance of the boroxine structure as cross-linkers. First, a hydrophilic copolymer poly(poly(ethylene glycol) methyl ether acrylate0.28m-co-glycidyl methacrylatem), abbreviated to poly(PEGMEA0.28m-co-GMAm), was synthesized using reversible addition-fragmentation chain transfer polymerization technique (Supplementary Fig. 23). The number average molecular weight (Mn) and polydispersity index (PDI) of the copolymer were determined to be 29, 800 g·mol− 1 and 1.58, respectively, by gel permeation chromatography (Supplementary Fig. 24). Subsequently, ethylenediamine was decorated to poly(PEGMEA0.28m-co-GMAm) through a ring-opening reaction with the epoxide moiety in GMA, which yielded poly(PEGMEA0.28m-co-AMAm) (Supplementary Fig. 23). Finally, poly(PEGMEA0.28m-co-AMAm) was functionalized with PBA and 2-methoxy phenylboronic acid (CH3O-PBA), respectively, through the Schiff-base reactions between amino group and aldehyde group, generating two corresponding copolymers, namely, poly(PEGMEA0.28m-co-AMAm-PBA0.57m) and poly(PEGMEA0.28m-co-AMAm-(CH3O-PBA)0.39m) (Supplementary Fig. 23). A third copolymer, poly(PEGMEA0.28m-co-AMAm-(HO-PBA)0.39m), was obtained by adopting BBr3 to convert the methoxy group of CH3O-PBA in poly(PEGMEA0.28m-co-AMAm-(CH3O-PBA)0.39m) into a hydroxyl group (Supplementary Fig. 23). The detailed characterization data of these polymers are described in Supplementary Fig. 25.
Poly(PEGMEA-co-AMA-PBA) and poly(PEGMEA-co-AMA-(CH3O-PBA)) are both water soluble due to the hydrophilic nature of poly(PEGMEA-co-AMA) and the favorable solubility of PBA and CH3O-PBA in water (Fig. 7b). Interestingly, after the methoxy group in CH3O-PBA of poly(PEGMEA-co-AMA-(CH3O-PBA)) was converted to the hydroxyl group, the resulted poly(PEGMEA-co-AMA-(HO-PBA)) formed a hydrogel when mixed with water, as shown in Fig. 7b.
The three copolymer/water mixtures were further analyzed using low-field nuclear magnetic resonance (LF–NMR) to measure the transverse spin-spin relaxation time (T2) of water in each mixture 48. The T2 distribution curve of each mixture displays three water components (Fig. 7c), indicating the presence of three forms of water with different degrees of freedom. The first component, T21 with the shortest relaxation time, corresponds to the protons of bound waters that are combined with the polymer molecules. The second component, T22, corresponds to protons of immobilized waters that are confined in the space between the polymer chains. The third component, T23 with the longest relaxation time, corresponds to protons of free waters49,50. In the poly(PEGMEA-co-AMA-PBA)/water and poly(PEGMEA-co-AMA-(CH3O-PBA))/water mixture, the T23 peak dominates the LF–NMR spectra with the proportionate peak areas of 80.3% and 65.5%, respectively (Supplementary Fig. 26). This indicates that water in these mixtures primarily exists as free water. In contrast, the T22 peak with proportionate peak area of 88.6% dominates the LF–NMR spectrum of poly(PEGMEA-co-AMA-(HO-PBA))/water mixture (Supplementary Fig. 26), which agrees with the well-established knowledge that water in the hydrogel is mainly confined within its 3D polymeric network.
The formation of poly(PEGMEA-co-AMA-(HO-PBA)) hydrogel was believed not to be driven by the noncovalent interactions within the copolymers, since hydrogels did not form by its counterparts poly(PEGMEA-co-AMA-PBA) and poly(PEGMEA-co-AMA-(CH3O-PBA)). Instead, the boroxine structures, which are derived from HO-PBA and crosslink the poly(PEGMEA-co-AMA-(HO-PBA)) chains, are responsible for the hydrogel formation. This presumption is supported by the fact that the hydrogel remains stable in both acidic (pH = 2) and alkaline (pH = 10) solution, as shown in Fig. 7b. This stability is consistent with the finding that the boroxine structure remains stable over a wide range of pH values (Fig. 4h). Moreover, the hydrogel could undergo a reversible gel–sol transition due to the dynamic nature of the boroxine structures. As shown in Fig. 7d, the hydrogel collapses in the presence of excess HO‑PBA trimer (left panel), which results from the de-crosslinking of the polymer networks via an exchange reaction of free HO‑PBA trimers with the boroxine cross-linkers (Fig. 7e). Removal of HO‑PBA trimers through washing with excess THF resulted in the reconstruction of the networks through the boroxine structures, thereby reforming the hydrogel (Fig. 7d, middle panel, Fig. 7e). Notably, the hydrogel was not influenced by the addition of excess PBA (Fig. 7d, right panel), which highlighted the role of HO-PBA. Overall, the poly(PEGMEA-co-AMA-(HO-PBA)) hydrogel represents the first boroxine-based hydrogel with high acid–base stability and reversible gel–sol transition, providing a new design strategy for hydrogel materials.
{kind=link}