3.1 Synthesis and surface modification of HSN and LP
To develop the degradable HSN for mitochondrial-targeted oligonucleotide delivery, first HSN were synthesized using a reverse microemulsion method,[26], and co-condensed with BTES (in-situ doping) to allow intracellular oligonucleotide release (Fig. 1a- top). BTES is a disulfide bridged siloxane that is reduced in GSH rich intracellular environments. Moreover, to enable NP tracing, a fluorescent dye-siloxane conjugate, RITC-APTES, was co-doped into the matrix of HSN. Synthesized HSN were uniform with a round morphology, hollow core, and mesopores as observed by TEM (Fig. 1b- left). Image J analysis of HSN revealed a diameter of 77.4 ± 4.2 nm (Fig. 1c). HSN were doped with 0.4 ± 0.1 µM RITC in 100 µg/mL and had a surface charge of 6.3 ± 3.2 mV, likely due to the presence of positive RITC-APTES on the surface (Fig. 1d-e, S1). Thus, here we developed highly fluorescent HSNs co-doped with BTES and RITC. Several groups have reported BTES doped etched-HSN for intracellular specific degradation,[27–28] however, we are the first to demonstrate BTES and RITC dual doped micro-emulsion-HSN.
LP reference NPs were synthesized by a modified dual-template stöber approach using ibuprofen as an auxiliary template and were doped with RITC-APTES (Fig. 1a- bottom).[29] To increase surface charge and improve oligonucleotide loading, amine groups were added at the surface by post-grafting (ex-situ surface modification) or by co-condensation with APTES ((3-Aminopropyl)triethoxysilane). The surface charge of post-grafted LP increased to 31.6 ± 12.6 mV, while doping resulted in LP with a surface charge of 8.9 ± 4.9 mV (LPD, Figure S2a). Although both LP were significantly more positive than LP without amine modification (LPC; -0.1 ± 5.6 mV, p < 0.0001, Fig. 1e, S2a), post-grafted LP were selected for further studies due to their higher surface charge. LP were spherical with dendritic large pores, a diameter of 116.1 ± 8.5 nm (Fig. 1b-c), and 0.6 ± 0.1 µM RITC per 100 µg/mL NP (Fig. 1d, S1).
We further investigated whether we could incorporate thiol groups in the LP core first by co-doping with MPTES ((3-Mercaptopropyl)trimethoxysilane) and then post-grafting with APTES to create LPSH. The resulting NP were positive (23.2 ± 4.7 mV, p < 0.0001, Figure S2a) and the presence of the amine and thiol groups was confirmed by reaction with a mixture of amine reactive FITC and thiol reactive ATTO-647 fluorescent dyes, forming tri-fluorescent LPSH (Figure S2b).
Although LP have been actively researched for over a decade as efficient silica-based drug delivery systems [30–31], we are the first to describe LP with a RITC-modified matrix and orthogonal surface (-NH2) and pore (-SH) functionalization. These LP can be functionalized both at the free amine and thiol groups using a variety of probes (e.g. fluorescent dyes, therapeutics, sensors) while the matrix remains fluorescent for real-time tracing (Figure S2b). This is a promising strategy to impart further functions to the NP such as (sub)cellular targeting, sensing, or imaging multimodality.[32] Overall, we successfully synthesized multifunctional LP and HSN.
3.3 Oligonucleotide incorporation and surface modification of HSN and LP
Next, oligonucleotides (LNA/DNA, DNA, PNA) were incorporated in HSN and LP nanoparticles (Fig. 2a, Table S1). To create HSN(LNA), a 33%LNA/67%DNA solution was added to the bulk oil phase, forming aqueous droplets of the reverse micro-emulsion, which were reaction centers for silica condensation (Fig. 2a- top). The encapsulation of different types of oligonucleotides was also investigated by replacing LNA/DNA solution with a DNA or PNA solution. We found that HSN were able to form in the presence of LNA/DNA (Fig. 2b-left) or DNA (Figure S3a) but not PNA (Figure S3b). The diameter or RITC signal of HSN(LNA) did not change compared to HSN (Figure S4, S1). However, the surface charge of HSN(LNA) was significantly higher than HSN (11.8 ± 5.3 mV, p < 0.01, Fig. 2c), possibly due to encapsulation of LNA/DNA causing positively charged RITC-APTES to migrate to HSN perimeters. The presence of LNA was confirmed by synthesis of HSN(LNA) with FITC-labelled LNA (LNA-FAM) resulting in green fluorescent NP (Fig. 2d). To confirm the encapsulation of LNA in the hollow core of HSN, LNA-FAM fluorescence and surface charge of HSN(LNA) was measured after washing 3 times. We observed that neither the fluorescence nor surface charge significantly changed (Figure S5). HSN have been developed as gene carrier systems in several studies.[33–35] However, in these approaches, oligonucleotides were complexed at the surface and not encapsulated in the hollow core, as we have done here. Our approach has the advantage that the HSN silica shell protects encapsulated oligonucleotides from degradation and enables HSN to be stored without the threat of oligonucleotide leakage.
To form LP(LNA), LP were immersed in LNA solution overnight, enabling active LNA loading in the pores by electrostatic absorption (Fig. 2a- bottom). Similarly to HSN, LNA incorporation in LP did not affect morphology, size, or RITC signal (Fig. 2b- right). However, in contrast to HSN(LNA), a drastic decrease in surface charge was observed from 31.6 ± 12.6 mV for LP to -19.5 ± 5.3 mV for LP(LNA) due to absorbed negative LNA masking the positively charged LP surface (p < 0.0001, Fig. 2c). Further, LNA-FAM absorption was highly dependent on NP type and surface charge; ~2-fold more LNA-FAM was incorporated in LP(LNA) compared to HSN(LNA), while LPC (LP doped with RITC without amine post grafting) absorbed approximately 10-fold less than LP (Fig. 2d, S6). The higher LNA incorporation in LP compared to HSN, is likely due to the increased surface charge and area of LP, allowing higher amounts of LNA to adsorb (Fig. 2d). Thus, surface charge and area are important factors to consider when designing NP for oligonucleotide delivery.
To facilitate intra-mitochondrial delivery, the surface of HSN and LP were modified with two different mitochondrial targeting moieties, MITO-porter and TPP (Fig. 2e). MITO-porter is a lipid bilayer composed of 1,2-dioleoyl-sn-glycero-3-phosphatidyl ethanolamine (DOPE), sphingomyelin (SM), and stearyl-octaarginine (STR-R8) in a molar ratio of 9:2:2 (Fig. 2e- orange, purple and red, respectively), which provides a positive charge, lipophilicity and enables cell uptake and endosomal escape.[36] Both TPP and MITO-porter enable mitochondrial membrane fusion and accumulation.[37] TPP was conjugated to the surface of HSN and LP via an amide bond to create HSNTPP and LPTPP while MITO-porter was surface adsorbed by the hydration method forming HSNM and LPM. LNA incorporated HSN and LP were also surface modified with TPP and MITO-porter, to form HSN(LNA)TPP, HSN(LNA)M, LP(LNA)TPP, and LP(LNA)M. Due to the highly positive nature of MITO-porter and TPP, the surface charge of HSNM and HSNTPP was significantly higher compared to non-modified HSN (25.6 ± 5.2 for HSNM and 46.3 ± 9.0 mV for HSNTPP) (p < 0.0001, Figure S7). This was also observed for HSN(LNA) with surface charges increasing to 15.6 ± 9.8 mV and 43.7 ± 5.4 mV for HSN(LNA)M and HSN(LNA)TPP, respectively (p < 0.0001, Fig. 2f). Similarly, TPP modification of LP also increased the surface charge (48.8 ± 11.8 mV, p < 0.0001, Figure S7). However, due to surface absorption of negative LNA, LPTPP surface charge drastically decreased (-33.8 ± 5.2 mV, Fig. 2f). Further, MITO-porter functionalization of LP did not affect the surface charge, while for LP(LNA), a drastic increase was observed to 31.6 ± 11.6 mV (Figure S7, 2f). To further confirm the presence of MITO-porter lipid bilayer, fluorescence analysis, and TEM imaging were used. For fluorescence analysis of HSNM and LPM, 2 mol% DOPE was replaced with the fluorescent lipid 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (18:1 (Δ9-Cis) PE) and co-localization of 18:1 (Δ9-Cis) PE signal at 488 nm and NP at 567 nm was observed (Figure S8). Moreover, TEM imaging revealed that the hollow core of HSNM was less visible and LPM reduced in porosity (Figure S9). To confirm TPP functionalization, Fourier-transform infrared spectroscopy (FTIR) was used. FTIR analysis of HSNTPP and LPTPP revealed emerging peaks at 700cm− 1 and 1650cm− 1 corresponding to deformation and stretching vibrations of phenyl C-H groups and amide C = O of TPP (Fig. 2g). The same peaks were also observed for LPTPP (Figure S10).
Here we showed that LP and HSN could be successfully surface functionalized with TPP and MITO-porter for mitochondrial targeting. Although several reports show MSN surface functionalization with lipid bilayers to improve colloidal stability[38] and reduce cargo leakage,[39] to the best of our knowledge, there are no reports on NPs functionalized with MITO-porter lipids.
3.4 LNA release and HSN degradation
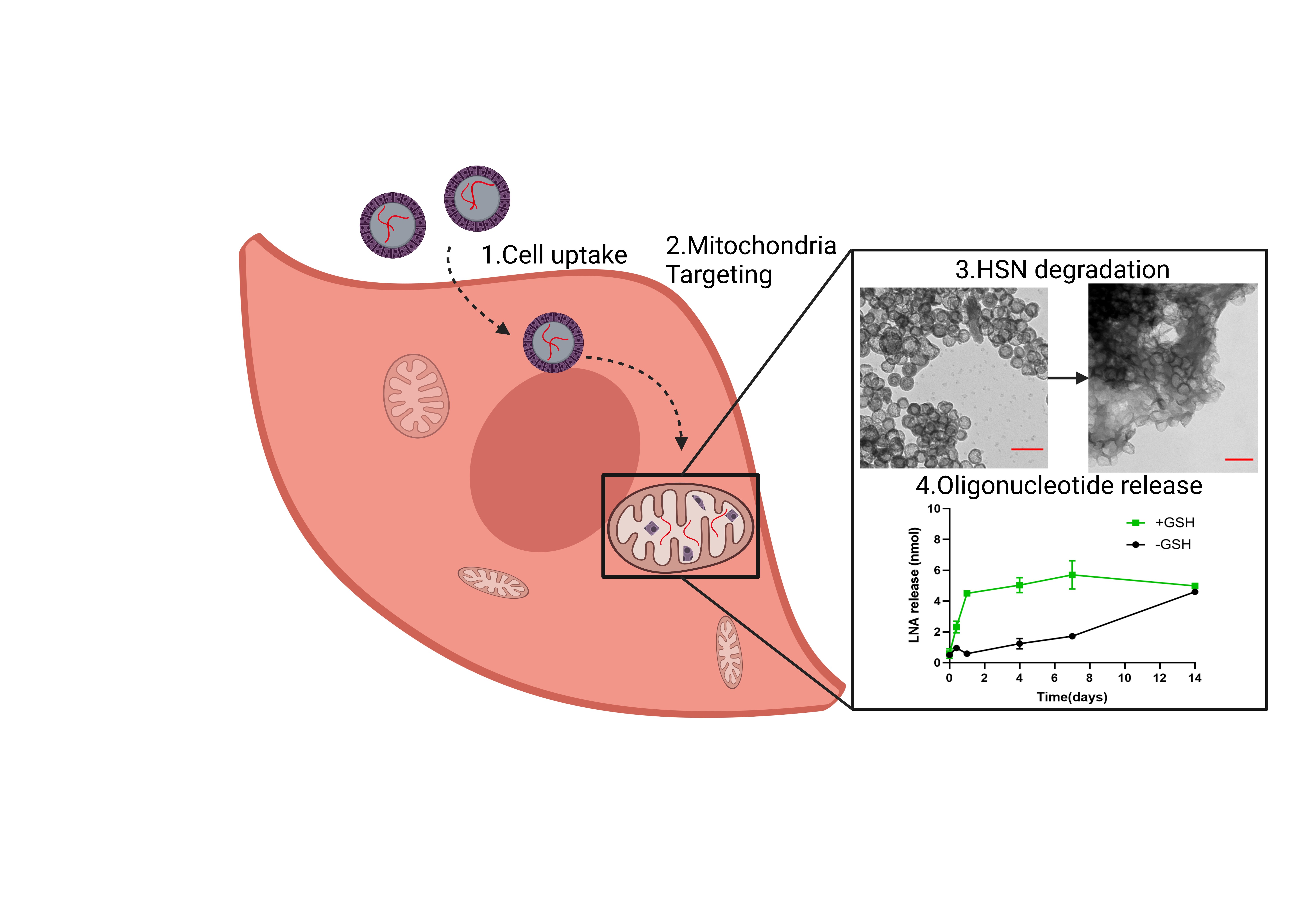
Next, LNA release profiles of HSN(LNA)TPP, HSN(LNA)M, LP(LNA)TPP, and LP(LNA)M in PBS or in 3 mM GSH (to mimic the intracellular GSH-rich environment) were measured over 14 days (Fig. 3a-b, d-e). Both HSN(LNA)TPP and HSN(LNA)M displayed faster LNA release when GSH was present. Specifically, 1 mg of HSN(LNA)TPP displayed gradual time-dependent LNA release from 0.6 ± 0.3 nmol at 0 days increasing at each time point to a maximum release of 5.0 ± 0.1 nmol at 7 days (p < 0.0001, Fig. 3a). However, when HSN(LNA)TPP was dispersed in PBS, LNA release was 4-fold less after 4 days compared to the release observed in the presence of GSH. LNA release kinetics from HSN(LNA), can be analyzed through an equation described by Higuchi et al., which takes into account the impact of the silica shell as a diffusion barrier.[25, 40] The Higuchi model is defined by the expression Q = KHt1/2, where KH is the Higuchi dissolution constant and refers to the rate constant of LNA diffusion through the silica shell. The calculated KH of LNA from HSN(LNA)TPP in PBS was 0.6, while KH in the presence of GSH was 4 times higher (KH = 2.4, Figure S11). Interestingly, for HSN(LNA)M, LNA release only began after 7 days in the presence of GSH and 14 days in neutral conditions, indicating that MITO-porter functionalization is capable of delaying LNA release by up to two weeks. By TEM analysis, we also found that HSN(LNA)TPP and HSN(LNA)M exhibited GSH-dependent degradation (Fig. 3c, S12), which could be correlated to the respective LNA release profiles (Fig. 3a-b, S11).
Overall, LP(LNA)TPP and LP(LNA)M exhibited faster LNA release than HSN(LNA). For LP(LNA)TPP maximum LNA release was already observed after 6 h reaching 6.7 ± 0.2 nmol in PBS which was indistinct from release in the presence of GSH (Fig. 3d). While for LP(LNA)M the release rate was 3.5 X faster in the presence of GSH and reached a maximum of 7.9 ± 0.4 nmol after 7 days (p < 0.0001, Fig. 3e) indicating that MITO-porter might be degraded by GSH. These findings suggest that MITO-porter is both responsive to GSH and acts as a gating system for LNA release from our HSN and LP.
Here we showed that both HSN(LNA) and LP(LNA) can efficiently release high amounts of LNA. Interestingly, despite being commonly used for gene delivery studies, oligonucleotide release kinetics from LP and HSN have not previously been reported, only oligonucleotide incorporation efficiencies were measured which were between 0.4 and 9 nmol oligonucleotides/mg.[10, 30–31, 33, 35, 41–42] In these studies, NP with relatively low oligonucleotide incorporation demonstrated transfection efficiencies comparable to established gene vectors such as Oligofectamine or Fugene.[43] For example, Zhan et al. described HSN with 0.8 nmol/mg pDNA transfected LoVo cells at efficiencies up to 48%.[33] While for LP with 2.5 nmol/mg siRNA a ~ 30% higher reduction in cancer cell viability was observed compared to oligofectamine.[10] Therefore, our HSN(LNA) and LP(LNA) that release up to 5.0 ± 0.1 nmol and 8.6 ± 0.1 nmol LNA/mg, respectively, should in theory, demonstrate comparable efficacy to what has previously been observed.
Further, we showed that HSN(LNA) and LP(LNA) display different release profiles depending on the presence or absence of GSH. Specifically, LP(LNA) exhibited fast LNA release throughout 6 h, indicating that quick NP uptake into the cell (< 6 h) is needed for their optimal use. Thus, LP could be used in endothelial or cancer cells that display NP uptake < 6 h.[44–45] In contrast, slow LNA release was observed from HSN(LNA) probably because the release was dependent on silica sphere degradation (Fig. 3a-c, S12). BTES doping of HSN allows oligonucleotides to be released upon degradation in intracellular conditions. This eliminates the need for laborious post-synthetic loading and gating modifications that are necessary for controlled HSN gene delivery.[34–35] Further, degradation of HSN enables cell elimination and clearance preventing side effects of NP accumulation, such as inflammation.[46] Significantly reduced HSN(LNA) degradation and cumulative release was observed in the absence of GSH, due to the lack of reduction of disulfide bonds in the HSN matrix. These results indicate that HSN(LNA) are relatively stable in extracellular spaces, lowering possible associated risks with premature, extracellular oligonucleotide release. Additionally, several studies report the use of GSH responsive etched-HSN to deliver drugs, such as DOX.[47–48] However, in these HSN only small molecules could be delivered, which were released within 1 day. Sustained, intracellular release of oligonucleotides as in our HSN(LNA) can help maintain an effective therapeutic dose in cells increasing efficacy and mitigating possible negative side effects.[49] For example, the gradual release of antisense oligonucleotides comparable to mtDNA turnover (~ 2 weeks) reduces the risk of homeostatic imbalance and cell death caused by blocking the replication of too many mtDNA.[50]
We also found that MITO-porter surface functionalization of LP(LNA) and HSN(LNA) resulted in delayed LNA release of 4 and 14 days, respectively. Delayed LNA release by MITO-porter functionalization could be a result of the charge stabilization of LNA with highly positive STR-R8 and steric hindrance of LNA passing through the lipid layer. Previous reports of lipid bilayer functionalized MSN showed delayed cargo release up to 48 h.[51–54] Moreover, MITO-porter functionalization of HSN(LNA) also showed reduced degradation (Figure S11).[55] We also observed that the rate of LNA release from LP(LNA)M was significantly increased in the presence of GSH. This is likely due to the degradation of MITO-porter in the intracellular environment as also observed for similar liposome formulations.[56] Thus, MITO-porter functionalization can be used as a novel tool for introducing intracellular responsivity and slowing LNA release which is especially important for gene therapy of mitochondrial diseases.
3.5 Biocompatibility and mitochondrial targeting
The biocompatibility and intracellular uptake of MITO-porter or TPP functionalized HSN and LP was assessed in MABs with an 80% m.3243A > G mutation load derived from an MM-patient, as potential target cells for mitochondrial delivery of oligonucleotides. MABs were exposed for 24 h to 10–500 µg/mL LPTPP, HSNTPP, LPM, and HSNM, and metabolic activity was assessed using the MTS assay. MABs metabolism was not affected upon exposure up to 100 µg/mL for any of the NPs tested. Concentrations above 200 µg/mL LPM resulted in decreased cell metabolism (p < 0.001, Fig. 4a). Next, flow cytometry was used to assess intracellular uptake as a function of MITO-porter and TPP surface functionalization. MITO-porter and TPP functionalization significantly increased cell uptake compared to un-functionalized HSN and LP (Fig. 4b). Specifically, 21.7 ± 2.3% of cells had taken up LP while 61.8 ± 6.1% and 75.7 ± 2.6% of MABs had internalized LPM or LPTPP, respectively (p < 0.0001, Fig. 4b- left). A similar trend was observed for HSN; 29.5 ± 0.7% of MABs took up HSN and 50.7 ± 3.2% contained HSNM. However, 31.4 ± 9.1% of MABs had internalized HSNTPP, which was indistinct from HSN uptake efficiency (p < 0.001, Fig. 4b- right). Fluorescence microscopy images revealed that HSN were mostly observed as extracellular intracellular aggregates, while some LP were observed in the cytoplasm (Figure S13). On the other hand, high intensities of MITO-porter and TPP functionalized NP were observed in intracellular locations.
The mitochondrial targeting ability of functionalized HSN and LP was assessed by fluorescence microscopy using MitoTracker deep red and TEM imaging after 24 h exposure (Fig. 5). Overlap of the MitoTracker channel with RITC-doped NP was analyzed qualitatively in merged channels (Fig. 5a) while mitochondrial co-localization was quantified by Pearson’s correlation coefficient (r) analysis of 6 microscopy images (Fig. 5b). Pearson’s correlation coefficient (r) is a measurement of linear correlation from − 1 (negative) to 1 (positive) which in this case refers to the spatial co-occurrence of NP fluorescence at 565 nm with mitochondria fluorescence at 647 nm (Fig. 5b).[57] We observed no correlation in the localization of bare HSNs with mitochondria since r was found to be 0.0 ± 0.1, while for bare LP a slight positive correlation was observed; 0.1 ± 0.1. For TPP and MITO-porter modified HSN there was a significant increase in mitochondrial co-localization to 0.4 ± 0.2 or 0.3 ± 0.1, respectively (Fig. 5b). While for LP, MITO-porter and TPP functionalization increased Pearson’s coefficient to the same extent (0.4 ± 0.2, Fig. 5b). Mitochondrial localization was also assessed by TEM imaging after 24 h exposure. We observed that HSN and LP without surface modification did not appear near mitochondria, while TPP and MITO-porter functionalized NP were mostly observed at mitochondria proximal locations (Fig. 6). Mitochondrial uptake of TPP functionalized NP could be observed for HSNTPP (Fig. 6). Further, LPTPP and HSNTPP could escape endosomes while LPM and HSNM remained mainly within endosomes (Figure S14).
Here we showed that TPP and MITO porter functionalized LP and HSN nanoparticles are biocompatible, and show increased cell-internalization and mitochondria targeting ability in patient derived MABs compared to non-functionalized HSN and LP. All NP formulations were non-toxic up to high concentrations of 100 µg/mL (Fig. 4a). For MITO-porter functionalized NP, we observed mild toxicity at concentrations above 100 µg/mL. Similar results were obtained by Kawamura et al. and is likely the effect of octa-arginine, which is known to become toxic at high concentrations.[58–59] We also showed that surface functionalization of HSN and LP with either TPP or MITO-porter led to enhanced cell uptake compared to non-functionalized HSN and LP. It has been well documented that the surface properties of NP have a drastic effect on the degree of cell uptake.[60–61] Specifically, it is known that positively charged, lipophilic NP surfaces lead to high cell uptake.[62–63] Thus, it is unsurprising that NP modification with positively charged lipid bilayer MITO-porter and positive, phenyl-rich TPP increases cell uptake. However, it is unclear why we observed a difference between TPP-functionalized HSN and LP.
Surface functionalization with TPP and MITO-porter also led to significantly increased mitochondrial localization compared to un-functionalized HSN and LP. Mitochondrial co-localization was quantified by Pearson’s correlation coefficient (r), which is deemed more accurate than other approaches since the calculation is based on mean intensity derivation.[57] We found that HSN were randomly distributed (r ~ 0.0) while LP were slightly co-localized with mitochondria (r ~ 0.1), probably a result of the higher surface charge of LP. High surface charge can lead to mitochondrial targeting as a result of the negative membrane potential of mitochondria.[64] However, when HSN and LP were functionalized with TPP and MITO-porter, Pearson’s coefficient significantly increased (r ~ 0.3–0.4). Although it has been suggested that only r > 0.5 indicates significant colocalization[57], artificially high r values can be easily created if background pixels are not eliminated[65], which is the case for most mitochondrial co-localization studies.[66–67] Here, we set color thresholds to the cell perimeter based on calcein, eliminating background. Pearson’s analysis also revealed that MITO-porter functionalized HSN targeted different mitochondria within cells most evenly; a feature important for homogeneous mitochondrial drug delivery.
TEM imaging confirmed that MITO-porter functionalized NP were internalized, but mostly trapped in endosomes (Figure S14). It is known that cell-penetrating peptide (CPP) octa-arginine promotes micropinocytosis leading to entrapment in macropinosomes.[68] However, CPP also stimulates escape from macropinosomes by membrane fusion.[68–70] As such, our observation indicates that endosomal escape likely occurs 24 h after cell uptake. In contrast, we showed that TPP functionalized NP were capable of endosomal escape within 24 h (Figure S14). TPP-mediated endosomal escape has been previously demonstrated and is likely a result of the proton sponge effect where intra-endosomal protonation of TPP causes an influx of chloride ions and leads to osmotic swelling and eventual endosomal rupture.[71] Our data indicate that TPP-mediated cell uptake and endosomal escape happens faster in comparison to MITO-porter, which is possibly due to TPP uptake occurring by endocytosis rather than macropinocytosis.[72] Nevertheless, both MITO-porter and TPP functionalized NP were located proximal to mitochondria as shown by TEM imaging. Mitochondrial internalization of NP has rarely been visualized however, TEM analysis is critical for making adequate conclusions about the intracellular location, status of degradation, endosomal entrapment and mitochondrial internalization of NP. With use of TEM imaging, we show that our HSN were capable of endosomal escape, located proximal to mitochondria and were not degraded prior to mitochondrial uptake enabling mitochondrial targeted gene delivery. Overall, we demonstrated that HSN and LP surface functionalized with MITO-porter and TPP were biocompatible, internalized into MABs and showed significant mitochondrial targeting ability.
{kind=link}