Based on the lack of knowledge that was identified concerning the effect and influence of the birthing room on the woman and her baby, and the likelihood that maternity wards in some countries will be in need of either reconstruction or new construction of labour wards, we identified the need to conduct a randomised controlled trial (RCT).
This study protocol (Version 1, 19th June 2019) is described in accordance with the SPIRIT checklist (Standard Protocol Items: Recommendations for Interventional Trials) [49]. For details see additional file 1. All major changes to the protocol (eg, changes to eligibility criteria, outcomes, analyses) will be notifed to the trial registry, ethics review board, investigators and participants.
Aim, hypothesis and study design
The overall purpose of this project is to extend the evidence-based knowledge on the design of birthing rooms and their influence on labour, birth and childbirth experiences in nulliparous women in active, spontaneous labour at ≥37 gestational weeks.
The hypothesis has been inspired by and built on: the mechanism of the effect of environment on birth [18-25] our own, unpublished systematic literature review, interviews with women some days after having given birth [50] aspects pinpointed by health care professionals at the study hospital and women from a user council (the agency Födelsehuset), information from the pilot study conducted in Canada [46], and from the ongoing study in Denmark [47].
Since the experience and influence of the healthcare environment is personal and complex, its effects have to be studied with objectively measurable methods as well as qualitative methods [7].
Our study is a randomised, controlled unblinded superiority trial (RCT), with two parallel groups comparing the effects of two types of birthing rooms. Care in a regular birthing room will be compared with care in an adaptable birthing room with a person-centred approach (“new room”). In this new birthing room, the woman can adapt physical aspects according to her own choices, thus creating an environment with a sense of safety, integrity and familiarity. Various outcomes of labour and birth, women´s self-reported experiences of the birthing room, childbirth and quality of life are measured. Furthermore costs are compared between the care in the ‘regular room’ and the “new room”. An additional ethnographic study will explore the influence and meaning of the birthing rooms (regular rooms and “new room”) on and for women giving birth.
Intervention
The study takes place at one of three labour wards at Sahlgrenska University Hospital (SUH), Gothenburg in the west of Sweden. The study takes into account a newly renovated birthing room, new technology, and education of staff to make best use of the updated facilities.
The labour ward serves women at >34 weeks of pregnancy. In 2018 the labour ward had 4237 births, where 1372 (32.4%) of the women were nulliparous with a single foetus in cephalic presentation at ≥37 weeks with spontaneous start of labour.
All birthing rooms are fully equipped with all necessary medico-technical devices. One of the regular birthing rooms has been reconstructed into a person-centered adaptable birthing room for the intervention group. This room provides the woman with more choices to adapt it according to her preferences. The main differences between the “new room” and the regular rooms are described in Table 1.
Recruitment and randomisation of participants
Eligible participants are women ≥18 years of age that are classified as “Robson 1” i.e.: nulliparous women at ≥37 gestational weeks, with a single live foetus in cephalic presentation, and in spontaneous labour [51]. They should understand either Swedish, English, Arabic or Somali or have access to an interpreter when necessary. When arriving to the labour ward they should be in the active phase of labour as defined in Sweden at the time of study, i.e. 2 of these 3 criteria fulfilled: spontaneous rupture of membranes; 2–3 painful contractions in 10 minutes; cervix dilated >3-4 cm or effaced and open >1 cm. Women with induced labour, planned caesarean section, multiple gestation or in the latent phase of labour will be excluded from the study.
All nulliparous women arriving to the labour ward are received in either a regular room or a so-called “assessment room”. The midwife clarifies if the woman is in active labour or not by checking the woman´s contractions, rupture of membranes, pain status, bleeding, fetal position by abdominal palpation and, in most cases, cervical status by vaginal examination. Auscultation of fetal heartbeat with Pinard’s stetoscope and recording of fetal heartbeat for 20-30 minutes with a Cardiotocography is also performed.
If both types of birthing rooms are vacant (i.e., the “new room” and one of the seven regular rooms), a woman fulfilling the inclusion criteria, and her companion, will be informed about the study orally and by written information, and will be invited to participate. The information comprises that:
- There are two types of birthing rooms that will be tested in the study: a specially designed room that has increased potential to be adapted to personal wishes and needs, and a regular birthing room.
- It is only the physical design that differs between the two rooms - the specially designed room will have the same level of medical safety and technology as the regular room.
- The responsible staff will explain the functions that are available in the room to which the woman will be allocated.
Since the midwife often needs time to clarify if the woman is in active labour or not, randomisation directly at time of arrival is usually not possible. Women who are admitted when the “new room” or one of the regular birthing rooms are not available, will not be included in the study. Women who choose not to participate in the study, will not be included in the study. The protocol is applied in exactly the same way for everyone; thus all women are treated equally.
A woman who wants to participate signs a written consent and is randomised to care in either the “new room” (intervention group), or to care in one of the regular rooms (control group). The randomisation system is managed by an independent statistician at an agency, who has prepared an allocation list based on randomly-generated numbers, placed the designated allocation in sequentially numbered, sealed envelopes kept in a study folder only available for staff members at the labour ward. To minimise the risk of detection bias, the woman will at randomisation be provided with the unique four-figured ID-code printed on the sealed envelope. The midwife opens the next sealed envelope containing details of the allocation, and informs the woman and her partner that they have been allocated to either the “new room” or the regular room. Blinding of either the attending staff or the participants is not possible. Midwives are not aware of the randomisation sequence and the envelopes are opaque. The independent statistician ensures that the randomisation procedure is followed.
The responsible midwife and other staff will follow the woman to the randomised room and provide care following the same care guidelines in both types of room. The enrollment and flow of the RCT is illustrated in Figure 1.
Any woman wishing to withdraw from the study, from either group, will be withdrawn immediately. However, without any persuasion from the researchers, she will be asked if she is willing for her data to remain in the database. If she agrees, her data will remain and be analysed with the rest of her allocated group’s data (i.e., intention to treat); if she disagrees, her data will be deleted. If a woman, randomised to the “new room”, wants to withdraw from the study at any time, she will be transferred and cared for in a regular room (i.e. standard care in that hospital). If a woman, randomised to a “regular room”, wants to withdraw from the study at any time, she will continue to be cared for in that room (i.e. standard care in that hospital).
Pilot testing
The study was pilot tested during December 2018, to test the study routines, identify and eliminate potential obstacles and simplify the implementation of the study. Before the start of the pilot study the staff at the labour ward received instruction on how to use the facilities in the room and the room was regularly used for the staff to feel comfortable within the environment. After that, nine women fulfilling the inclusion criteria were included in the pilot study. The pilot test showed that the study procedure was feasible, some information sheets were revised to be more understandable, and a new checklist was developed to facilitate identification of eligible women. Furthermore, the inclusion criteria were further specified regarding that the included woman must be in active labour at enrollment and not spend several hours in a regular room before randomisation since this could affect the experience of the birthing environment negatively.
Data collection and outcome measures
The primary efficacy endpoint is a composite score, measured before discharge from the hospital and compared between the two groups. To ensure holistic data for assessing the effect of the room on labour and birth outcome both medical and experiential outcomes are included in the score. The composite score is 1 if all the following four parts of the composite variable are fulfilled and 0 otherwise:
- No use of synthetic oxytocin for augmentation of labour
- Spontaneous vaginal births (i.e. no vaginal instrumental birth or CS)
- Normal postpartum hemorrhage (i.e. bleeding <1000 ml.)
- A positive overall childbirth experience: 7-10 on a Likert scale with anchor 1=bad, 10=good
The secondary efficacy outcomes that will be measured and compared between the two groups are: All four primary variables, Body Mass Index (BMI), gestational week at birth, seeking help for fear of childbirth, mental illness treatment, rupture of membranes, amniotomy, use of synthetic oxytocin for augmentation of labour: duration and highest dose, duration of labour, duration of active labour, use of bathtub, use of epidural anaesthesia, maternal fever during labour, duration of pushing stage, position of woman for vaginal birth, episiotomy, mode of birth (vaginal spontanoeus, vacuum extraction and CS), indication for instrumental vaginal birth and CS, cervical tear, perineal tears divided in grades, postpartum haemorrhage (>1,000 ml.), manual removal of placenta, stillbirth, neonatal death during hospital stay, Apgar score at 5 minutes (1 to 10, and grouped < 4 and < 7), umbilical cord blood samples at birth, sex of newborn, birth weight, skin to skin in the first hour, breastfeeding within first two hours, admission to the neonatal unit, and length of mother’s hospital stay. Self-reported experiences and outcomes will be measured 2 hours after birth, after discharge, after 3 and 12 months with: specific questions concering: experience of the room, childbirth ecperience as a whole, Childbirth Experience Questionnaire (CEQ) [52], Fear of Birth Scale (FOBS) [53], Fear of giving birth again [54], and health-related quality of life using the EuroQol 5 Dimension (EQ-5D) health state questionnaire [55]. If the woman does not answer the 3 or 12 month questionaires she will get up to 3 reminders once a week, the first two reminders by email followed by one phone text message. Using such techniques in our previous studies results in response rates of 70-80%.
Data entry: The researchers are blinded to the participants´ responses since the ID-code is used during data entry. The woman´s self-reported data will be registered two hours after birth (touch screen), and 3- and 12 months after birth (Web questionnaire or paper questionnaire). Data from the medical record will be entered by a research midwife in a web-based form. This data will be double checked. The only one of the four parts of the primary composite outcomes that could potentially be swayed is postpartum haemorrhage. To guard against any possibility of bias the following routines are established:
- the blood loss will be measured, not estimated;
- the same team of midwives will be caring for women in both arms of the study, and for all other women not in the study, so a biased individual will have little opportunity to care for women in the “new” room very often;
- there are at least two healthcare personnel present at any birth, so both would need to be willing to falsify the results for this to happen, which is unlikely to occur with any frequency;
- the researchers make checks on all data and will be on the lookout for any potential anomalies.
All electronic data will be kept on a password-protected computer in a locked office at the labour ward for the study. All hard copies of participant details will be kept in a fireproof, locked cabinet for research data at the University of Gothenburg. All data will be stored for ten years after end of study, and then destroyed securely.
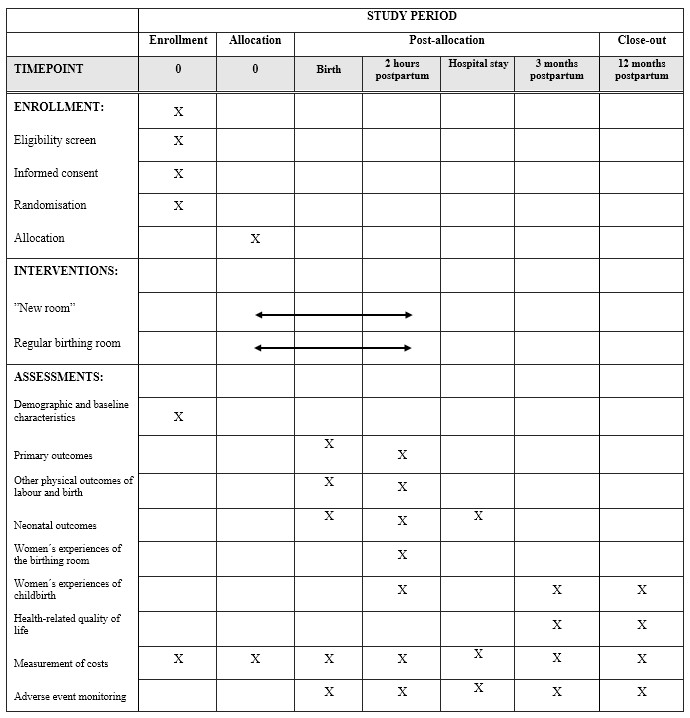
The healthcare costs related to the hospital stay will be collected from the hospital administrative records. As the study population constitutes only of nulliparous women at term without severe increased risks, we assume that adjustments for comorbidity are not needed. A schedule of enrollment, interventions and assessments is presented in Table 2.
Sample size: A total of 1274 women are required (637 per group), to detect a reasonable clinical difference in the primary composite outcome of 8% between the groups (45% in control vs. 53% intervention), with two-sided Fisher´s test, 80% power and 5% significance level using ‘intention to treat’ population. To allow for 10% attrition 1401 women are required. This sample size calculation is based on data from the labour ward for the target group (Robson 1) in the year 2017 in which the three first parts in the composite score were fulfilled in 47.9% of the target group. Among these, based on a national register study [56] as no data were available from SUH, we assumed that 94% could have a positive ‘overall childbirth experience’. This implies that 0.479*0.94 = 0.450 =45% fulfils all four parts in the main composite outcome. Data from the Swedish Pregnancy Register show that 84.1 % of all registered women had a positive childbirth experience in 2015-2016 [56].
Statistical methods
All main analyses will be performed on the intention-to-treat (ITT) population and followed according to randomisation. The primary analysis and selected secondary efficacy analyses will also be performed on the ‘per protocol’ population. For comparison between the two randomised groups Fisher’s non-parametric permutation test will be used for continuous variables, Mantel-Haenszel Chi2-test for ordered categorical variables and Fisher’s exact test for dichotomous variables and Chi2-test for non-ordered categorical variables. For dichotomous outcome variables two-sided 95% confidence intervals (CI) for the difference in proportions between groups will be calculated as well as risk ratios (RR) with 95% CI. For continuous outcomes two-sided 95% CI for the difference in means between groups will be calculated based on Fisher’s non-parametric permutation test. The main analysis will be the above unadjusted analyses.
We do our primary analysis with the composite primary outcome. If there are group imbalances in important baseline characteristics found, complementary analyses adjusted for these variables will be performed with analyses of covariance for continuous variables and with generalised linear models with binomial distribution and log link function in order to calculate adjusted RRs with 95% CI for dichotomous variables. The distribution of the variables will be given as mean, Standard Deviation (SD), median Quartile 1 and 3 (Q1, Q3) for continuous variables and as numbers and percentages for categorical variables. Imputations for missing data will be performed, when applicable. Missing data will be imputed using fully conditional multiple imputation in the main analysis. A sensitivity analysis will be performed with the full analysis dataset without imputation. All tests will be two-sided and conducted at the 5% significance level.
The ethnographic study
An ethnographic study with participant observations documented by field notes [57], will be conducted in both types of birthing room during day, evening and night shifts. This study will explore the influence and meaning of the birthing rooms on women giving birth and thus increase the understanding of how healthcare contexts and human interactions can affect birth outcomes. It also takes an outsider perspective when interpreting how environments and cultures affect us, such as how a birthing room influences the birthing women.
The exact number of observations will be determined by data saturation, i.e. when sufficient variation and quality of data to answer the study aim are achieved. During the observations, there will be short, clarifying talks with the observed women. Field notes will also contain the observing researcher’s reflections. Based on preliminary analysis of observations, clarifying in-depth interviews will be conducted (around 6 weeks after birth), with some of the women, at a place chosen by the woman. Field notes of observation, reflection notes, recorded spontaneous talks, and follow up interviews are transcribed verbatim for analysis. The goal for the data analysis will be to explore and describe the influence of the birthing environment on the women giving birth in the two types of room. Data will be analysed using interpretation of the women´s experiences, conceptions, imaginations, and practices in the birthing context in relation to safety, familiarity and choice. To ensure validity, further analyses of primary interpretations will be conducted by the responsible researcher in collaboration with the research team through recurrent discussions. This process is guided by following the hermeneutic spiral movement between the whole, the parts, and a new whole described in categories and themes [58].
Roles and responsibilities
The research team consists of the Steering Committee (Marie Berg, Lisa Goldkuhl, Christina Nilsson Helle Wijk and Cecily Begley), who are responsible for the quality and conduct of the study, and the wider members of the team (Steering Committee together with Hanna Gyllensten, Göran Lindahl, Kerstin Uvnäs Moberg). Given the intervention (use of different type of birthing room, where care in labour is unchanged), no adverse events are envisaged. However, a Data and Safety Monitoring Committee (DSMC) will be established for the project, with clinicians, a service user representative and a statistician. Information leaflets for women include details of how to make complaints or report adverse incidents. Any such incidents reported to the team will be passed on to the DSMC for consideration. This group will be provided with an account of project progress annually, and will be contacted if there are any complaints or adverse incidents attributed to the trial. They will conduct an objective interim analysis on data from the first half of the trial, to assess for any possible harms, and will provide an outside opinion on the safety of the intervention, at end of trial. We have not set any stopping guidelines for futility as it is important to continue to full recruitment in order to have sufficient numbers to assess results in all groups.
{kind=link}