Aliphatic esters are highly valuable products and chemical intermediates. They are present in a broad range of important biologically active molecules (Fig. 1a)1-3 and among the most versatile intermediates in the step-economical and orthogonal synthesis of aliphatic acids, aliphatic amides, aldehydes, ketones and alcohols. Traditionally, synthesis of aliphatic esters relies on the esterification of carboxylic acids, anhydrides or acyl chlorides with alcohols. These approaches need the pre-installation of a carboxyl group in the substrate. A complementary and more versatile alternative can use alkenes as starting materials, which are readily available and abundant petrochemical feedstock starting materials and synthetic intermediates.4 Alkoxycarbonylation reactions of alkenes, developed by Reppe in the 1950s, is the most significant industrial process under transition-metal catalysis (Fig. 1b).5 Based on the palladium-catalyzed alkoxycarbonylation reaction of ethylene, the current most advanced industrial process (Lucie Alpha process) to methyl propionate is used on an industrial scale to produce more than 300000 tons of products annually.6 Although transition-metal catalyzed alkoxycarbonylation reactions are powerful tools to synthesize esters, these processes rely on the use of high pressure of toxic and flammable CO at high temperature, which often require specific equipment and safety precautions.7 Additionally, application of these transformations is limited by the challenges associated with the regioselective alkoxycarbonylation of olefins as well as the harsh reaction conditions. Usually, a mixture of linear esters and branched esters is afforded.7 Thus, direct catalytic and regioselective synthesis of aliphatic ester derivatives from unactivated olefins under mild conditions remains an unsolved challenge in modern synthetic chemistry. It is clear that a novel and efficient approach is needed to address the above issues in the regioselective alkoxycarbonylation process.
As shown in Figure 1d, we expected that a free radical based method involving the addition of an alkoxycabonyl radical to olefin might be an attractive and alternative strategy. With this strategy, the alkoxycarbonylation of olefins will provide the desirable linear esters. In general, alkoxycarbonyl radicals are generated most commonly from the corresponding diethyl azodicarboxylate,8 selenides9 and xanthates10. Additionally, alkoxycarbonyl radicals can be formed from carbazates by treatment with metal catalysts and stoichiometric quantities of oxidants.11 Recently, it was reported that alkoxycarbonyl radicals could be produced by photoredox-catalyzed fragmentation of methyl N-phthalimidoyl oxalates (Fig. 1c).12 These existing methods for generating alkoxycarbonyl radicals from alcohols require multistep synthetic procedures. Moreover, most of the reported examples dealt with structurally simple alkoxycarbonyl radicals, while only a few reports exploiting complex alkoxycarbonyl radicals was described.13
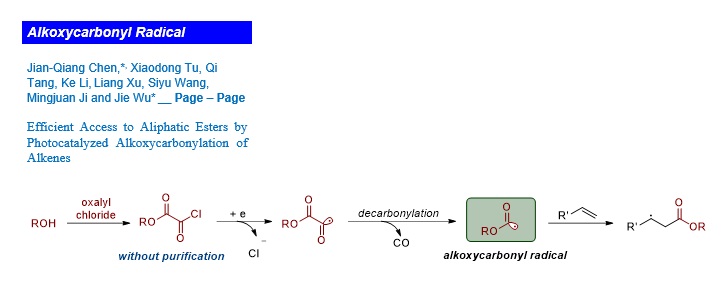
It is well known that single-electron reduction of aroyl and sulfonyl chlorides by photocatalyst would provide aroyl14,15 and sulfonyl radicals,16 respectively. Prompted by these results, we envisioned that it might be possible to identify a novel strategy for the generation of alkoxycarbonyl radicals from the corresponding acyl chloride via photoredox catalysis (Fig. 1d). A photocatalytic strategy to introduce both the desired ester group and a versatile electrophile at the β-position of ester group would be quite useful in the preparation of significant compounds, due to the complementary reactivity of the esters. To the best of our knowledge, the selective alkoxycarbonylchlorination of alkenes leading to β-chloro esters from alkyloxalyl chlorides has not been explored yet.
To verify the feasibility of this free-radical alkoxycarbonylchlorination strategy, ethyl chloroformate (2a’) was used as the alkoxycarbonyl radical source. Unfortunately, none of the target product 3a was obtained when a mixture of 4-vinyl-1,1'-biphenyl (1a), ethyl chloroformate (2a’), 2,6-lutidine and Ir(ppy)3 in acetonitrile at 40 oC was irradiated with blue LEDs for 24 h. (Table 1, entry 1). The failure of this result might be rationalized by the difficulty of promoting single-electron reduction of chloroformate because of its lower reduction potential. After careful investigation, we found that the readily available and inexpensive ethyl chlorooxoacetate (2a)17 was an ideal alkoxycarbonyl radical precursor. This chlorooxoacetate might be readily reduced by the excited state of photocatalyst Ir(ppy)3 to generate alkoxycarbonyl radical through CO-extrusion. As shown in entry 2 (Table 1), the linear product 3a was observed,18 accompanied by the appearance of a byproduct 6a (10% yield) when ethyl chlorooxoacetate (2a) was used instead of ethyl chloroformate (2a’). This transformation proceeded with excellent regioselectivity and gave rise to the direct formation of linear ester 3a (the branch isomer could not be found by H-NMR) under the operationally simple conditions. Additionally, examination of photosensitizers revealed that Ru(bpy)3Cl2 produced a better yield (entry 4), however, the reaction took much longer time for completion. To further improve the yield of alkoxycarbonylchlorination product of ethyl 3-([1,1'-biphenyl]-4-yl)-3-chloropropanoate (3a), the reaction temperature was increased and the yield was slightly increased (to 66%, entry 5 in Table 1). Evaluation of different solvents showed that acetonitrile was the best choice in this transformation (entries 5−7). Notable, α,β-unsaturated ester compound 6a was the major product when DMF was used instead of acetonitrile. Interestingly, decreasing the amount of 2,6-lutidine led to a significantly improved yield and substrate (1a) was fully consumed (entry 8). As expected, visible light irradiation and photosensitizer were essential components for this alkoxycarbonylchlorination reaction (entries 9−10).
With the optimized conditions in hands, the generality of this alkoxycarbonylchlorination reaction was then evaluated. As outlined in Table 2, a wide range of styrenes were efficiently workable in this alkoxycarbonylchlorination protocol. For example, electron-neutral and electron-rich styrenes were all suitable substrates (3b−3d, 63−68% yield). This photocatalyzed alkoxycarbonylchlorination strategy was effective as well for electron-deficient styrenes, as demonstrated by the installation of fluoro, chloro, bromo, ester and aldehyde groups (3e−3i, 41−75% yield). Furthermore, para-chloromethyl styrene, which could be further functionalized through nucleophilic substitution reaction, gave rise to the target product (3j) in good yield. Moreover, 1- and 2-vinylnaphthalenes were found to be competent substrates (3k and 3l, 63% and 83% yield, respectively). The efficiency of this process was not impeded by ortho-, meta-methyl or fluorine substitutions on the aromatic rings (3m−3p, 40−63% yield). A natural-product-derived styrene substrate was also coupled with high level of efficiency (3q, 62% yield). This result further demonstrated the potential of employing native functionality to access novel structural analogues and to provide the further functionalization.
Having demonstrated the capacity of activated alkenes in this alkoxycarbonylchlorination reaction, we next investigated the conversion of various alkyloxalyl chlorides. As shown in Table 2, the yield was decreased to 38% (3b vs 3r) when commercially available methyl oxalyl chloride was used as the alkoxycarbonyl radical precursor in the reaction with styrene. We applied this strategy to the derivatization of alcohol-containing biologically active molecules. By treatment of the corresponding alcohols with oxalyl chloride in dry DCM, alkyloxalyl chlorides generated in situ were employed directly in the photoredox protocol after removal of the excess amount of oxalyl chloride and DCM by vacuum distillation.19 Alkyloxalyl chlorides derived from primary and secondary alcohols were found to be generically successful in this alkoxycarbonylchlorination reaction. Chiral amino alcohol derivative reacted with styrene leading to product 3s in 64% yield. Additionally, this transformation was insensitive to steric hindrance around the site of alkoxycarbonyl radical (3t−3w, 45−87% yield). For example, alkyloxalyl chloride derived from (-)-borneol provided the desired product (3t) in 87% yield. Notably, chlorooxoacetates derived from other nature products including androsterone (product 3u, 45% yield), β-cholestanol (product 3v, 69% yield) and menthol (product 3w, 68% yield), were workable as well.
Encouraged by the results described above, we subsequently explored the transformation of various unactivated alkenes. The reaction of non-conjugated alkene (but-3-en-1-ylbenzene 4a) failed to provide the corresponding product under the optimal conditions (Table 3, entry 1). However, the formation of desired β-chloro ester product 5a was observed when Ir(ppy)3 was employed (Table 3, entry 2). A better result was obtained when the reaction was performed at 30 oC (Table 3, entry 3, 44% yield). By raising the equivalents of photosensitizer Ir(ppy)3 and the alkoxycabonyl radical source 2a, the yield of this reaction was increased from 44% to 74% (entries 4−7).
On the basis of the above optimized conditions, we next explored the reaction scope with diverse unactivated alkenes. As illustrated in Table 4, a wide range of unactivated alkenes coupled with chlorooxoacetate 2a efficiently, giving rise to the corresponding products. Transformation of long-chain α-olefins produced the desired compounds 5b−5e in reasonable yields (53−69%) with complete regiocontrol. Remarkably, hydrolysis of ethyl β-chlorooctanoate 5b promoted by HCl would provide an antibacterial compound.3 Cetylates and stearates are privileged motifs encountered across the molecular sciences,20 particularly in food chemistry,21 medicinal chemistry,22 flavor and fragrance industry.23 Reaction of 1-pentadecene or 1-heptadecene would afford the addition product of ethyl β-chlorocetylate 5d or ethyl β-chlorostearate 5e in moderate yields, respectively. Moreover, a versatile electrophile at the β-position of ester would certainly accelerate the synthesis and discovery of new bioactive molecules. More nucleophilic 1,1-disubstituted alkenes could smoothly participate in this transformation, affording the corresponding product 5f in 78% yield. It was found that a striking feature of this reaction was the exclusive formation of β-chloro esters without any undesired rearranged products, even in cases where benzyl, cyclohexyl and tert-butyl were present in the α-position relative to the double bonds. Aside from various carbon scaffolds, some functional groups were found to be tolerated under these conditions, such as esters (5j and 5k), ketones (5l and 5m) and amide (5o). Excitingly, a tri-substituted alkene of cholesterol reacted cleanly to provide the corresponding product 5n in an unoptimized 21% yield and with excellent diastereo- (> 20:1 dr) and regioselectivity. Furthermore, electron-deficient olefin was also entirely converted into the expected β-chloro product 5o. In all cases, only one regioisomer was obtained, making this reaction fully regioselective.
Since 1-tetralone moiety is widely found in the core structure of natural products,24 organic synthetic intermediates25 and bioactive compounds,26,27 much attention has focused on the synthesis of 1-tetralone derivatives. Therefore, the above method was applied to the preparation of 1-tetralone derivatives. As shown in Table 5, several 1-tetralone derivatives (5’) were prepared in moderate yields through a radical alkoxycarbonylation/cyclization with 1-arylpent-4-en-1-ones under the same reaction conditions. This convenient method described above will be potentially useful for the synthesis of bioactive compounds containing 1-tetralone moiety.
Late-stage carbon-hydrogen bonds functionalization of pharmacologically active compounds is a remarkable strategy for the discovery of novel functional compounds because it avoids laborious de novo construction of new analogues, increases the efficiency of structure-activity relationship investigation, and provides new candidates that might have never been explored.28 With this in mind, we envisioned that direct alkoxycarbonylchlorination of alkenes gave rise to the corresponding β-chloro esters, which could then undergo HCl elimination upon workup to give α,β-unsaturated esters.29 This formal β-selective alkenyl C-H alkoxycarbonylation strategy is quite appealing. Additionally, α,β-unsaturated esters are key components of synthetic building blocks, pharmaceuticals and natural products.30-32 Thus, the synthesis of α,β-unsaturated esters remains an actual interesting task in the context of the development of improved synthetic methodologies.33 As illustrated in Table 6, by treatment of alkenes 1 and ethyl chlorooxoacetate 2a under the optimal conditions, followed by elimination with excess amount of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) at room temperature for 30 min, α,β-unsaturated esters 6 were obtained with high levels of chemo-, region- and stereoselectivity. A wide range of olefins could participate in this transformation, affording the corresponding α,β-unsaturated esters. Various electron-neutral, electron-rich and electron-poor styrenes were viable substrates (6a−6j, 47−75% yield). 1,1-Disubstituted aryl alkenes reacted with ethyl chlorooxoacetate 2a giving rise to the corresponding tri-substituted α,β-unsaturated esters in moderate yields (6k−6l, 54−59% yield). Reaction of 3,4,5-trimethoxystyrene could produce the desired cinnamic ester derivative 6m. To demonstrate the amenability of this alkoxycarbonylation process to late-stage application, bexarotene34 analogue was subjected to the standard conditions and the corresponding alkoxycarbonylation product 6n was obtained in 57% yield.
We next explored the generality of the decarbonylative alkoxycarbonylation/cyclization protocol. As shown in Table 7, It was found that a broad range of N-arylacrylamide derivatives and alkyloxalyl chlorides were suitable substrates in this transformation. A number of N-arylacrylamides bearing both electron-rich and electron-poor substituents in the aromatic ring, underwent alkoxycarbonylation/cyclization with ethyl chlorooxoacetate 2a leading to the corresponding products in good to excellent yields. Notably, reaction of antetrahydroquinoline or tetrahydrobenz-azepine derivative afforded the desired tricyclic product 8e or 8f in 91% and 90% yield, respectively. Furthermore, N-arylacrylamide with a naphthalene substituent on the nitrogen reacted with ethyl chlorooxoacetate 2a smoothly, providing the corresponding oxindole-3-acetate 8l in 83% yield. Reactions of N-arylacrylamides with ethyl, isopropyl and benzyl-protecting groups on the nitrogen atom proceeded well, affording the substituted products in good yields (8m−8o, 72−80% yield). Additionally, acrylamide with a benzyl group at the α-position could convert into the desired product 8p in 91% yield. To demonstrated the practicability of this photoredox process, a gram-scale experiment was carried out. which provided the corresponding oxindole-3-acetate 8a in 77% yield (1.53 g). However, transformations of unprotected N-arylacrylamide, monosubstituted olefin (R3 = H) and indoline derivative were not successful.
Subsequently, reactions of various alkyloxalyl chlorides were investigated under the standard conditions. The yield of the alkoxycarbonylation/cyclization product was slightly diminished (8q vs 8a) when methyl oxalyl chloride was used instead of ethyl chlorooxoacetate 2a. Alkyloxalyl chlorides derived from primary alcohols were found to be successful in this conversion. For example, reaction of alkyloxalyl chloride derived from 4-phenyl-1-butanol provided the desired product in quantitative yield (8r, 99% yield). The long-chain alkyloxalyl chloride derivative was also an effective alkoxycarbonyl radical source, and the corresponding product 8s was furnished in 85% yield. To further demonstrate the advantage of this conversion, we applied this strategy to the derivatization of alcohol-containing biologically active molecules. A series of chiral secondary alcohols were examined, and the representative examples are shown in Table 7. It’s noteworthy that the corresponding chiral secondary alkoxycarbonyl radical intermediates could readily convert into the desired products without any decarboxylated products (8t−8y, 58−84% yields).27 Additionally, the reaction was insensitive to steric hindrance around the site of alkoxycarbonyl radicals (8t−8v, 61−84% yield). The transformations proceeded efficiently as well when β-cholestanol (product 8w, 81% yield), androsterone (product 8x, 81% yield) and cholesterol-derived (product 8y, 58% yield) alkyloxalyl chlorides were used. Excellent diastereo-selectivity (> 20:1 dr) was observed for β-cholestanol substrate. Chiral amino alcohol derivatives could also be applied to the synthesis of oxindole-3-acetates (8z−8ad, 67−82% yield). These experiments demonstrated that this strategy was compatible with the functionalization of biologically active molecules bearing polar functional groups (8x and 8z−8ad). However, transformations of benzyl oxalyl monochloride and tertiary alcohol derivative were not successful.
Furoindoline moiety is broadly found in the core structure of biologically active compounds and natural products.35-42 Considering that oxindole-3-acetates are versatile building blocks for constructing heterocycle-fused indolines,43,44 we decided to apply the above method to the preparation of furoindoline derivatives. As shown in Table 3, by treatment of N-arylacrylamides 7 and ethyl chlorooxoacetate 2a under the optimal conditions, followed by reduction with LiAlH4 at 0 oC, furoindolines 9 were obtained as expected with excellent diastereoselectivities.43 This route was highly efficient, and a range of furoindoline derivatives was readily produced in only two steps from simple precursors. Additionally, N-(4-methoxyphenyl)-N-methylmethacrylamide reacted with ethyl chlorooxoacetate 2a under the standard conditions, giving rise to the desired tricyclic furoindoline 9m, which could be readily converted into bioactive alkaloid physovenine in two steps.35,36 In contrast to previous reports for the synthesis of physovenine, including Sharpless epoxidation,37 Grignard reaction,38 Diels–Alder reaction,39 catalytic asymmetric Heck reaction,40 intramolecular Michael addition,41 [3,3]-sigmatropic rearrangement,42 this method was better in the atom-and step-economic.
We next applied this methodology to the concise synthesis of expensive dihydronaphthalene derivative,45 which is important precursor for a wide range of biorelevant molecules.46 In contrast to the conventional routes,47,48 our method not only decreased the step count, but also simplified the operation greatly (Fig. 2). Furthermore, this strategy was utilized in the formal synthesis of marketed drug ozagrel, which was an antiplatelet drug and marketed in Japan in 1989.49-51 As shown in Figure 2, this alkoxycarbonylation reaction enabled us to access key compound 6i, which was subsequently subjected to the substitution reaction with imidazole providing product 10i in 70% yield. An additional hydrolysis of compound 10i would afford ozagrel.51
As mentioned above, both photosensitizer and visible light were necessary for this alkoxycarbonylation process. Therefore, on the basis of previous reports52 and the experimental results, a plausible mechanistic pathway was proposed in Figure 3. Irradiation of photosensitizer Ir(ppy)3 (16) would produce a long-lived (τ = 1.9 μs) excited photocatalyst *Ir(ppy)3 (17), which was a strong single electron donor (E1/2 [Ir(ppy)3+/*Ir(ppy)3] = -1.73 V vs. SCE in MeCN)53 to reduce the reactant. Ethyl chlorooxoacetate (2a) would accept an electron from *Ir(ppy)3 (17) and then produce acyl radical intermediate (12). This acyl radical intermediate (12) would undergo decarbonylation leading to the alkoxycarbonyl radical 13,54 The driving force for this decarbonylation reaction was mainly the extrusion of stable carbon monoxide and the generation of stable alkoxycarbonyl radical which could react with alkene to afford carbon radical intermediate 14. In the meantime, a single electron transfer from *Ir(ppy)3 to compund 2a would generate the oxidized Ir(ppy)3+ (18), which could be reduced by alkyl radical 14 to provide the ground state Ir(ppy)3 and cation intermediate 15. This cation intermediate 15 would be attacked by chloride ion to form the desirable β-chloro ester 5 (path A). The formation of intermediates of type 15 was difficult in the case of electron-poor olefins (product 5o). A competitive chain pathway might not be excluded (path B). The carbon radical 14 might abstract chlorine atom from ethyl chlorooxoacetate (2a) to produce the target product 5 and regenerate acyl radical intermediate 12. To verify the above hypothesis, a radical trapping experiment was performed with the addition of TEMPO. As a result, the related alkoxycarbonyl-TEMPO product was confirmed through GC-MS (for details, see Supporting Information) This observation demonstrated that this transformation might proceed through a radical process.
{kind=link}