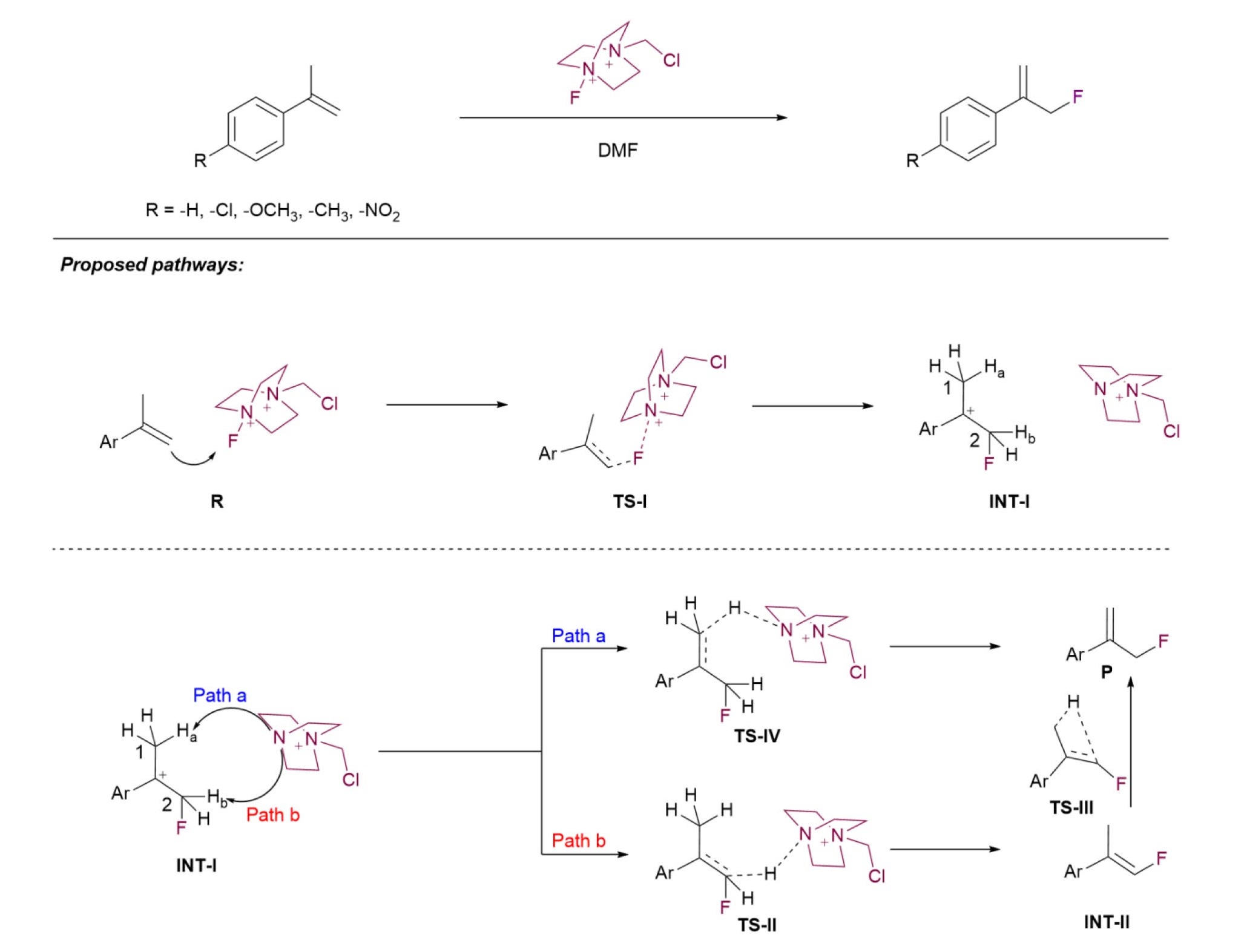
The reaction that was studied involved the direct electrophilic fluorination of alkenes to form aryl allyl fluorides in a fast and efficient manner, without the need of any transition metal catalyst.[24] The reaction employed styrenes as olefin source, Selectfluor[25] as electrophilic fluorinating reagent and DMF as a solvent that furnished the allyl fluoride products in good to excellent yields. Lately, a similar reaction on acyclic alkenes have also been reported.[26] The proposed mechanism for allylic fluorination reaction is shown in Scheme 1. The first step of the proposed mechanism involves addition of electrophilic fluorine across the double bond of a styrene to form a tertiary carbocation intermediate (INT-1) via transition state (TS1). The elimination of hydrogen from INT-I can take place by two different pathways. The first pathway involves abstraction of hydrogen (Ha) from C-1 carbon to form the desired allylic fluoride product via transition state TS-IV, whereas the second pathway would involve the abstraction of acidic hydrogen from fluorine containing carbon C-2 to form vinyl fluoride intermediate INT-II via transition state TS-II, which can undergo rearrangement via transition state TS-III in order to furnish the desired allyl fluoride product P.
In order to understand the reaction mechanism and get insight into the desired reaction pathway, the reaction of α-methyl styrene with Selectfluor was chosen as a model reaction. Firstly, the reaction of α-methyl styrene with Selectfluor for the synthesis of (3-fluoroprop-1-en-2-yl)benzene (P) was investigated in gas phase at B3LYP/6-31G(d,p) level of theory. All structures along the reaction pathway as shown in Scheme 1 were optimized without any geometrical constrain and are shown in Fig. 2. The free energy profiles along with reaction coordinates for both pathways is shown in Fig. 3.
Gas phase calculations reveal that in the first transition state, labelled as TS-I, terminal carbon of alkene abstracts electrophilic fluorine from Selectfluor which leads to C2 – F (2.52 Å) bond formation and simultaneous bond cleavage of N – F bond (from 1.39 to 1.51 Å) as shown in Fig. 2. This can be confirmed from transition vector corresponding to the imaginary frequency of 238.21i cm-1. The energy barrier to reach TS-I from reactant R is 4.43 kcal/mol and it leads to the formation of a cationic intermediate INT-I which is -68.31 kcal/mol stable than the reactant (Fig. 3). The high stability of intermediate INT-I can be attributed to the weak hydrogen bonding interactions, and can be confirmed by the C2 – F----H and C2 – H----N bond distance of 2.49 Å and 2.60 Å, respectively. This intermediate can undergo two possible routes; path a and path b to form the desired allylic fluoride product as shown in Scheme 1. The path b involves the abstraction of C2 – Hb hydrogen that would lead to a TS-II which is 52.02 kcal/mol lower than the reactant (Fig. 3) and forms the vinyl fluoride intermediate INT-II (Scheme 1). In order to form the product, the vinyl fluoride intermediate has to go through the highly energetic concerted rearrangement pathway. The energy barrier to reach the TS-III from intermediate INT-II is 56.82 kcal/mol, which seems difficult to achieve under the reaction conditions (Fig. 3). On the other hand, path a involves direct abstraction of C1 – Ha hydrogen that would lead to TS-IV which is 14.9 Kcal/mol higher than the intermediate INT-1 (Fig. 3). Although abstraction of Hb hydrogen is as favorable as the abstraction of kinetically feasible Ha hydrogen due to the presence of electron withdrawing fluorine atom that decreases the pka; the high energy requirement of the transition state TS-III involving concerted rearrangement makes this pathway unlikely for the formation of product P.
Thus, the gas phase investigation of the reaction between α-methyl styrene with selectfluor indicates that reaction would proceed by the formation of transition state (TS-I) which further gives intermediate (INT-I) that leads to product formation via transition state TS-IV. We have further explore the effect of polar aprotic and polar protic solvent over the energies of reactants, intermediates, transition states and product involved in reaction mechanism. Firstly, the reaction of α-methyl styrene with Selectfluor was studied in polar aprotic solvent DMF at B3LYP/6-31G(d,p) level of theory. The energy profile of various species involved in solvent DMF is shown in Fig. 4. The energy of transition state TS-I was found to be 5.94 kcal/mol, which is higher than the gas phase reaction by an amount of 1.51 kcal/mol. Use of polar aprotic solvent DMF results in high destabilization of INT-I as compared to INT-II and P, which is obvious as carbocation intermediated are high energy species compared to the neutral molecules. However, drastic stabilization of TS-IV was observed as it is -41.10 Kcal/mol stable than the reactant, which is a difference of around 13.31 kcal/mol compared to the gas phase. Also, large energy difference between TS-III and TS-IV (50.04 kcal/mol), points towards the same conclusion that the highly reactive intermediate INT-I would prefer elimination from C1 rather than C2, to form the product in a single step rather than undergoing two step high energy barrier (INT-II→TS-III, 66.28 Kcal/mol) pathway. Therefore, we propose that the product formation from the intermediate INT-I will take place in a single step via path a.
Furthermore, in order to understand the effect of protic polar solvent on the reaction mechanism, we investigated the reaction of α-methyl styrene with Selectfluor in methanol using DFT calculations at B3LYP/6-31G(d,p) level of theory. The energy profile of various species involved in solvent methanol is shown in Fig. 5. The use of polar protic solvent results in destabilization of transition state TS-I by an amount of 2.18 kcal/mol compared to aprotic solvent DMF, while by an amount of 3.69 kcal/mol compared to the gas phase reaction. Additionally, it also destabilizes the intermediates INT1 (-43.50 Kcal/mol), transition state TSIV (-39.07 kcal/mol) and product P (-55.62 kcal/mol) as compared to DMF. These results indicates that the most preferred solvent for this reaction is polar aprotic solvent DMF which is in line with experimental results.[24]
In order to investigate the effect of electron-withdrawing and electron-releasing substituents on phenyl ring of α-methyl styrene on the energy requirement of the reaction. Substrates containing -Cl, -OCH3, -CH3 and -NO2 groups present over para position of phenyl ring of α-methyl styrene were chosen for DFT calculations in gas phase. The free energy profiles for reaction pathways for these substrates are shown in Fig. 6.
The reaction between 1-chloro-4-(prop-1-en-2-yl)benzene with Selectfluor is investigated using DFT at B3LYP/6–31 G(d,p) level of theory in gas phase. The results of DFT calculations shows that transition state (TS-1) for reaction of 1-chloro-4-(prop-1-en-2-yl)benzene with Selectfluor have energy requirement of 4.66 Kcal/mol which is higher than that required for unsubstituted substrate α-methyl styrene. This can be attributed due to electron-withdrawing tendency of the chlorine atom that destabilize the tertiary carbocation formed during the reaction. Similarly, the use of 1-methyl-4-(prop-1-en-2-yl)benzene and 1-methoxy-4-(prop-1-en-2-yl)benzene as olefin substrates with Selectfluor have energy requirement of 3.88 kcal/mol and 3.67 kcal/mol, respectively, which is lower than that required for unsubstituted substrate α-methyl styrene. Therefore, electron-releasing group increases the rate of reaction by stabilizing the carbocation intermediate as compare to electron-withdrawing groups. In case of substrate containing a strong electron-withdrawing group such as 1-nitro-4-(prop-1-en-2-yl)benzene, the transition state could not be resolved, which indicates that the reaction may not be feasible with this substrate. The order of reactivity of these substrates can be further justified by using frontier molecular orbital theory. According to FMO theory, the rate of a reaction depends upon the nucleophilic and electrophilic strength of the reagents which in turn is dependent on the energy difference between the HOMO and LUMO orbitals of these reagents.[27] Reactant with high energy HOMO can easily donate electron pair and act as good nucleophile while the one with low energy LUMO can easily accepts electron and can act as better electrophile.[28] The FMO calculations shows that HOMO is located on α-methyl styrene moiety with energy levels of -11.384 eV, -11.897 eV, and − 12.035 eV for substrates containing methoxy, methyl, and chloro groups, respectively, which indicates maximum reactivity for substrate containing methoxy group followed by substrates containing methyl and chloro groups. We further investigated the stability of transition states based upon HOMO-LUMO energy gap. It is always desirable to have lower HOMO-LUMO energy gap which points towards more stability of the transition states. In this context, the energy gap for the transition states (TS-1) for substrates α-methyl styrene, 1-chloro-4-(prop-1-en-2-yl)benzene, 1-methyl-4-(prop-1-en-2-yl)benzene and 1-methoxy-4-(prop-1-en-2-yl)benzene are shown in Fig. 7. The energy gap is minimum for 2.013 eV in case of methoxy substituent indicating its highest stability followed by methyl substituent (2.128 eV) and chloro substituent (2.243 eV).
{kind=link}