In the current study, we investigate the role of Ppp2r5d in the pathogenesis of DCM. We demonstrated that Ppp2r5d knockdown in cardiomyocytes and stimulation of cardiomyocytes with β-adrenoreceptor agonist ISO reduced mtStat3 levels, decreased the ETC complex activity, reduced the production, elevated the intracellular ROS levels, and exacerbated myocardial apoptosis. Moreover, Ppp2r5d knockdown in cardiomyocytes impaired the expression of hypertrophic markers and IL6 in response to ISO stimulation. Interestingly, myocardial-specific Ppp2r5d deficient mice exhibited an abnormal DCM-related phenotype showing enlarged ventricles, abnormal cardiac dysfunction, and accelerated ventricular remodelling. In addition, exacerbated mitochondrial damage in the DCM mouse model was observed. The present findings provide evidence that Ppp2r5d plays an important role in the pathogenesis of DCM.
Several processes such as inflammation, viral infection, and chemotherapeutic agents contribute to the development of DCM that ultimately leads to ventricular enlargement and HF. DCM is manifested by enlarged ventricles and impaired cardiac contractility in response to different causal stressors. Recently, we found elevated levels of the Ppp2r5d mRNA in the peripheral plasma of patients with DCM.2 This abnormal expression of Ppp2r5d mRNA was also observed in the heart tissue of DCM mice, which motivated us to further explore the potential role of Ppp2r5d in DCM. It has been reported that the B56δ protein, encoded by Ppp2r5d, regulates numerous critical biological processes such as DNA replication, and mitosis via distinct signal transduction pathways.27, 28 The primary form of PP2A is a heterodimeric trimeric complex consisting of a catalytic C subunit, a scaffold A subunit, and a regulatory B subunit. The regulatory B subunits comprise a large group of genetically and structurally diverse proteins that fall into four categories, B/B55, B′/B56, B′′/PR72, and B′′′/striatins. In mammals, B′/B56 contains five spliceosomes: B56α, B56β, B56γ B56δ, B56ε. Previous studies of B56δ were focused on developmental disorder syndromes such as mental retardation, epilepsy, macrocephaly, and developmental delay.4, 29Ppp2r5d knockdown promotes tumorigenesis and malignant development by enhancing phosphorylation at the Ser9 site of GSK-3β and the Ser62 site of the proto-oncogene C-myc.3 A recent study reported that the B56δ E420K mutation led to an overactive AKT-mTOR signalling pathway by regulating AKT, GSK3α, and S6 hyperphosphorylation, resulting in an uncoordinated cell proliferation.29 Another study also revealed that inhibition of tumorigenesis can be achieved through the PP2A/GSK3β/MCL-1 pathway. Under hypoglycemia/metformin treatment, upregulated Ppp2r5d dephosphorylated GSK-3β, leading to a decline in MCL-1 and cell death.30 ISO stimulation enhanced PP2A activity by upregulating B56δ Ser573 phosphorylation and subsequently by elevated intracellular phosphorylation of cTnI at Ser22/23 and cMyBP-C at Ser282 in cardiomyocytes.7 These findings give strong evidence for a potential function of Ppp2r5d in cardiomyopathy, although the mechanism remains unclear.
As we mentioned previously, our microarray data showed that the Ppp2r5d mRNA level is significantly altered in the peripheral blood of DCM patients. Moreover, our results revealed that the Ppp2r5d mRNA level is significantly downregulated in the cardiac tissue of DCM mice, but not in MI mice. Surprisingly, the microarray results showed that Ppp2r5d deficiency resulted in highly enriched differential expressed genes participating in pathways associated with hypertrophic and DCM. These results gave further evidence that Ppp2r5d is associated with DCM.
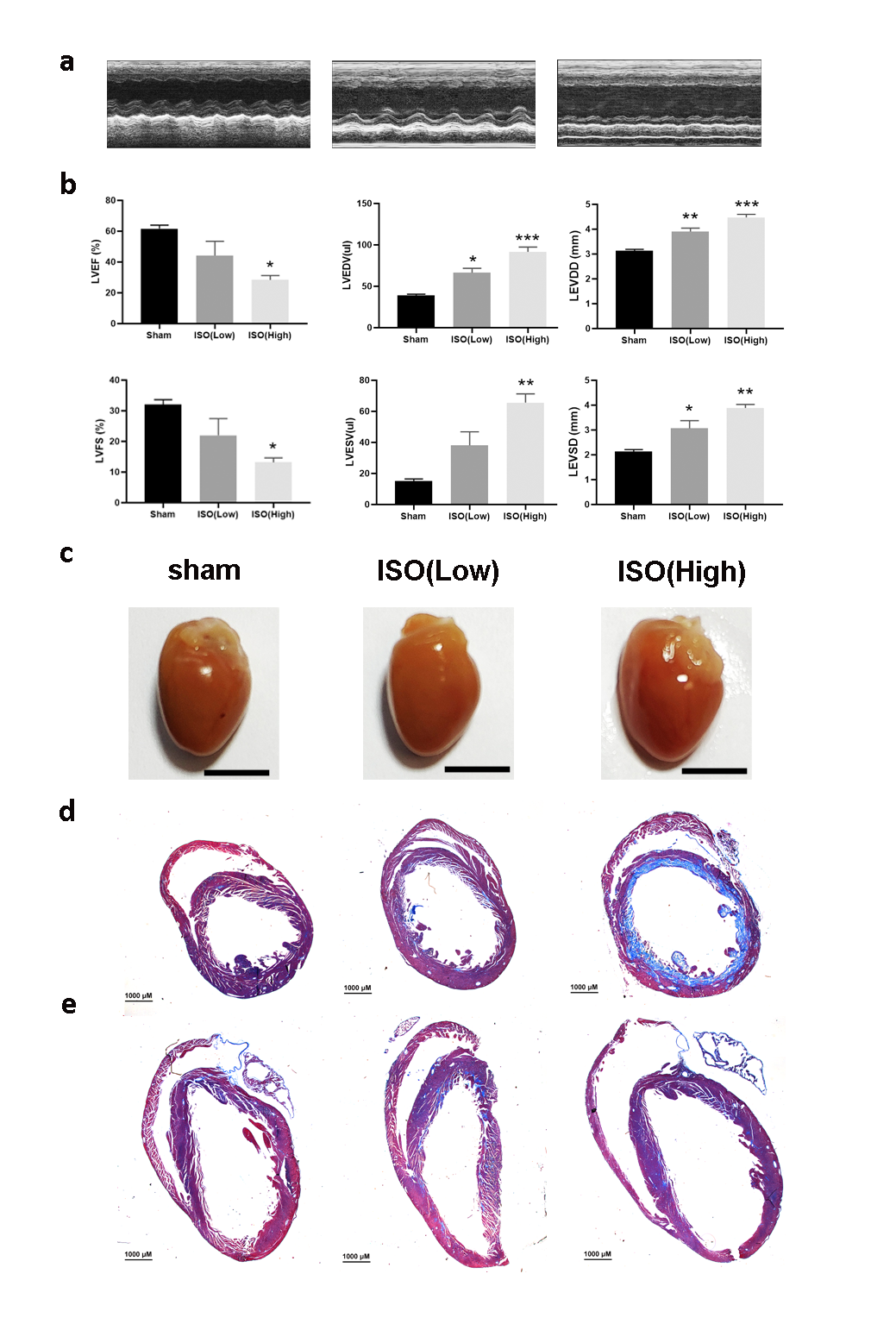
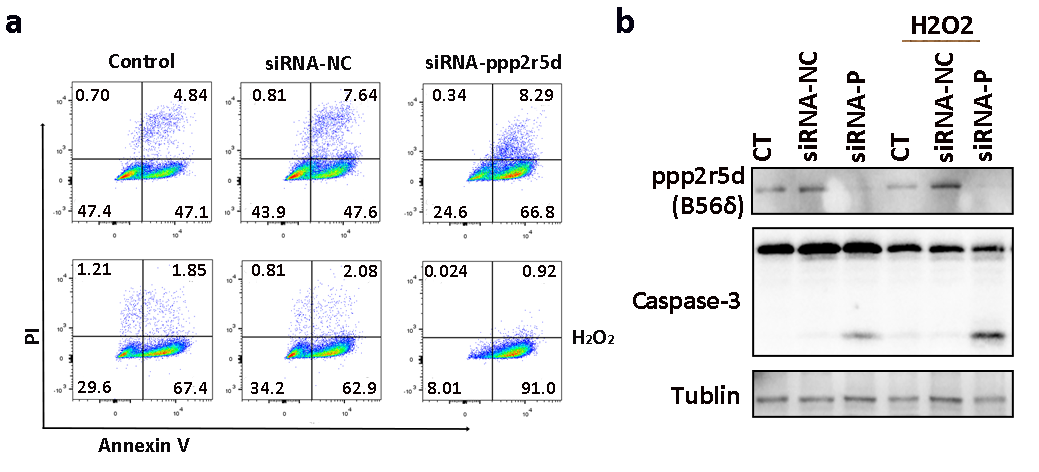
Next, we found that Ppp2r5d deficient MCMs are more sensitive to the detrimental cellular effects of the ISO- and H2O2-induced ROS. Intracellular ROS levels were significantly upregulated in the Ppp2r5d knockdown group as compared to the control group. Indeed, an excess of ROS is associated with mitochondrial dysfunctions and DCM is associated with oxidative stress and inflammation.31 Our results also demonstrated that the Ppp2r5d deficiency contributes to the reduction of intracellular ATP levels in non-stimulated and ISO-stimulated cells. We further revealed that the baseline mitochondrial respiration, ATP production, and maximal oxygen consumption were considerably reduced in the Ppp2r5d deficient MCMs as compared with the control cells under ISO stimulation. In vivo experiments, DCM mice which were treated with AAV-shRNA‐Ppp2r5d displayed severe cardiac dysfunction, ventricular fibrosis, a ventricular dilatation in comparison to the vehicle control group. Additionally, the ventricles of the Ppp2r5d deficient DCM mice indicated significantly more myofilaments and mitochondrial damages than those of the ventricles of the vehicle-treated DCM mice. Based on these results, we postulated that Ppp2r5d deficiency aggravates cardiac injury by promoting mitochondrial dysfunction. Therefore, we tested the expression levels of genes associated with ETC and ATP synthesis. During ISO treatment, the mRNA levels of Ndufv1, Sdhb, Cyc1, Uqcrc2, and Atp5a1, as well as the NdufB8 and Sdhb protein levels, were down-regulated in the Ppp2r5d knockdown MCMs. Moreover, the activity of the ETC complex I was impaired in the Ppp2r5d deficient MCMs after ISO stimulation.
Stat3 belongs to the STAT protein family and can be phosphorylated at two sites. For instance, the S727 site phosphorylation drives Stat3 into mitochondria (mtStat3) and is strictly related to mitochondrial electron transport chain activity.32–34 In this context, it was reported that mtStat3 increases the activity of ETC complexes I and II.18 Moreover, phosphorylation of the Y705 site drives Stat3 into the nucleus in which it acts as a transcriptional activator, which may be involved in the pathophysiology of cardiomyopathy. This hypothesis is strongly supported by the observation that inhibiting Stat3 Y705 site phosphorylation in a rat model of stress-induced cardiac hypertrophy significantly improved cardiac function.35 We also found that Stat3 phosphorylation levels were altered. Our data confirmed an increase in STAT3 Tyr705 phosphorylation and a decrease in Stat3 Ser727 phosphorylation in the Ppp2r5d deficient group, treated with ISO. The phosphorylation of Stat3 might induce mitochondrial dysfunctions and increase the expression of IL6 in cardiomyocytes. IL-6 contributes to the development of interstitial fibrosis in DCM.
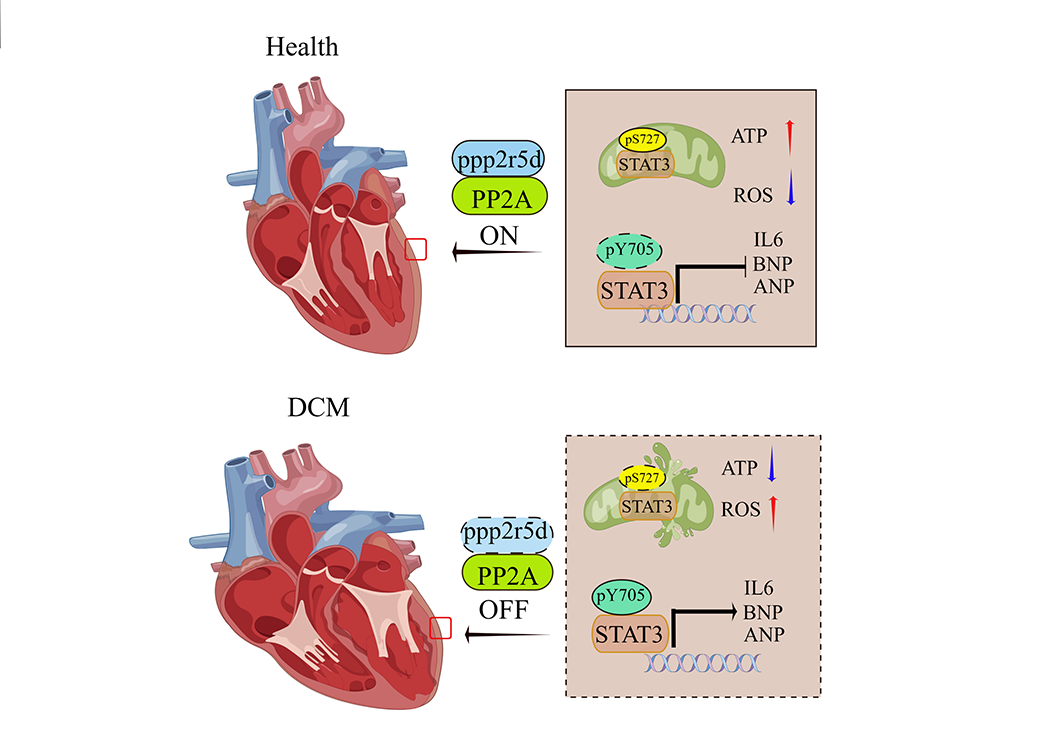
Collectively, these data suggest that Ppp2r5d is involved in the development of DCM and may represent a turning point in the elucidation of the DCM pathogenesis. A possible mechanism is presented in Fig. S4. We hypothesize that Ppp2r5d regulates the Stat3 phosphorylation to maintain mitochondria depending on energy homeostasis in normal functional myocardial tissue. Ppp2r5d is downregulated in cardiomyocytes during DCM development, which promotes mitochondrial dysfunction, and apoptosis in cardiomyocytes as well as aggravates myocardial fibrosis and ventricular remodelling.
However, although we identified the Ppp2r5d-dependent axis controlling the intact function of cardiomyocytes which is dysbalanced by the DCM pathogenesis, there are still open questions that should be answered in the future. It is not clear whether Ppp2r5d alone or via interactions with other cellular compounds modulates the Stat3 phosphorylation level. Finally, rescue experiments are needed to restore the function of Ppp2r5d in deficient pp2r5d cardiomyocytes thereby delaying the progression of DCM.
{kind=link}
{kind=link}
{kind=link}
{kind=link}