MICAL1 is an important monooxygenase that participates in regulating cytoskeletal rearrangement(Vitali et al., 2016; Wu et al., 2018; Zucchini et al., 2011). Recently, the potential widespread function of MICAL1 on cancer pathophysiological processes has attracted increasing attention. Studies have indicated that in experimental models, for example, MICAL1 promotes the malignant development of breast tumors(McGarry et al., 2021), MICAL1 has been perceived as being markers for renal clear cell adenocarcinoma(Yang et al., 2022). Our previous study showed that hypoxia induced MICAL1 expression in gastric cancer(Zhao et al., 2019). In this study, we revealed a new link between PlexinA1 and MICAL1 in positive regulation of gastric cancer cell migration, that is PlexinA1 maintains MICAL1 expression by Rac1/ROS dependent manner, thereby enhancing vimentin expression and cancer cell migratory ability.
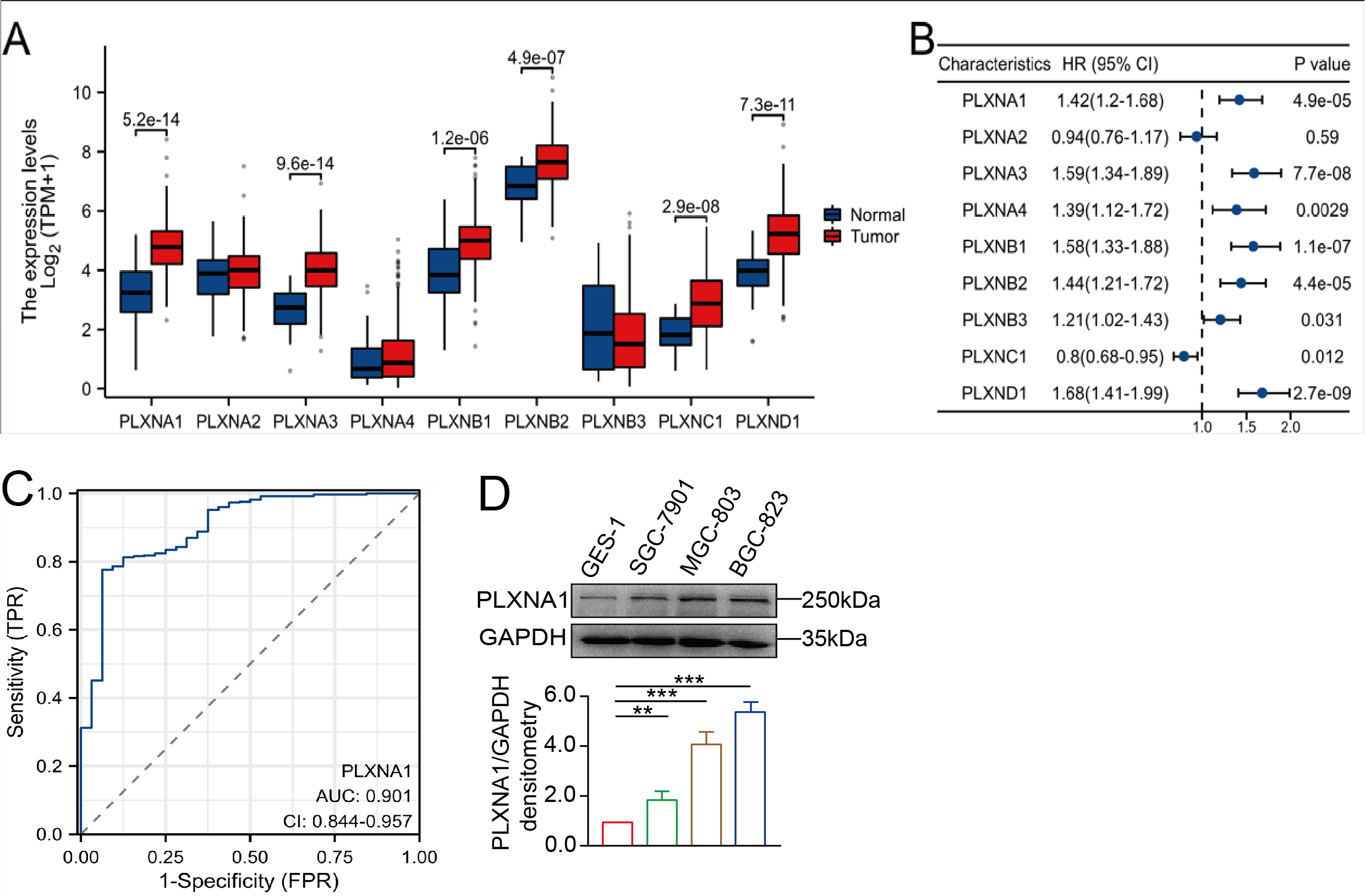
The results in this study showed high MICAL1 expression in gastric cancer tissues, and increased expression of MICAL1 was correlated with poor prognosis, indicating a close relation between MICAL1 and gastric cancer progression. The most novel finding was that the regulation of MICAL1 expression was strongly dependent on PlexinA1. MICAL1 was identified as an effector that took part in PlexinA1 signaling pathway for axon guidance(Jahan et al., 2022). MICAL1 can also mediate sema3a-induced F-actin collapse, thereby regulating shape changes of podocyte in diabetic mice(Aggarwal et al., 2015). Recently, the role of PlexinA1 in tumor progression has gained increasing interest from researchers. Pro-oncogenic effects of PlexinA1 were noted in certain tumors such as brain and gastric (Jacob et al., 2016; Lu et al., 2017). In the present study, we found that gastric cancer cell lines or patient samples expressed higher PlexinA1 than normal samples, and that tumors with high PlexinA1-expression are correlated with worse-survival rates. This oncogenic effect of PlexinA1 in gastric cancer was consistent with previous results from Yamada et al., who reported that PlexinA1 positively related with mRNA expression levels of proliferation-marker genes and lung cancer cell proliferation(Yamada et al., 2016). Consistent with the report that PlexinA1 is required for neural crest migration(Wagner et al., 2010), our findings revealed that PlexinA1 accelerates gastric cancer cell migration by stabilizing MICAL1 expression. Interestingly, no obvious alteration in MICAL1 mRNA levels were found after silencing of PlexinA1, suggesting that PlexinA1 might not change MICAL1 expression at transcriptional level. Then, we focused on revealing the mechanisms through which PlexinA1-induced stabilization of MICAL1.
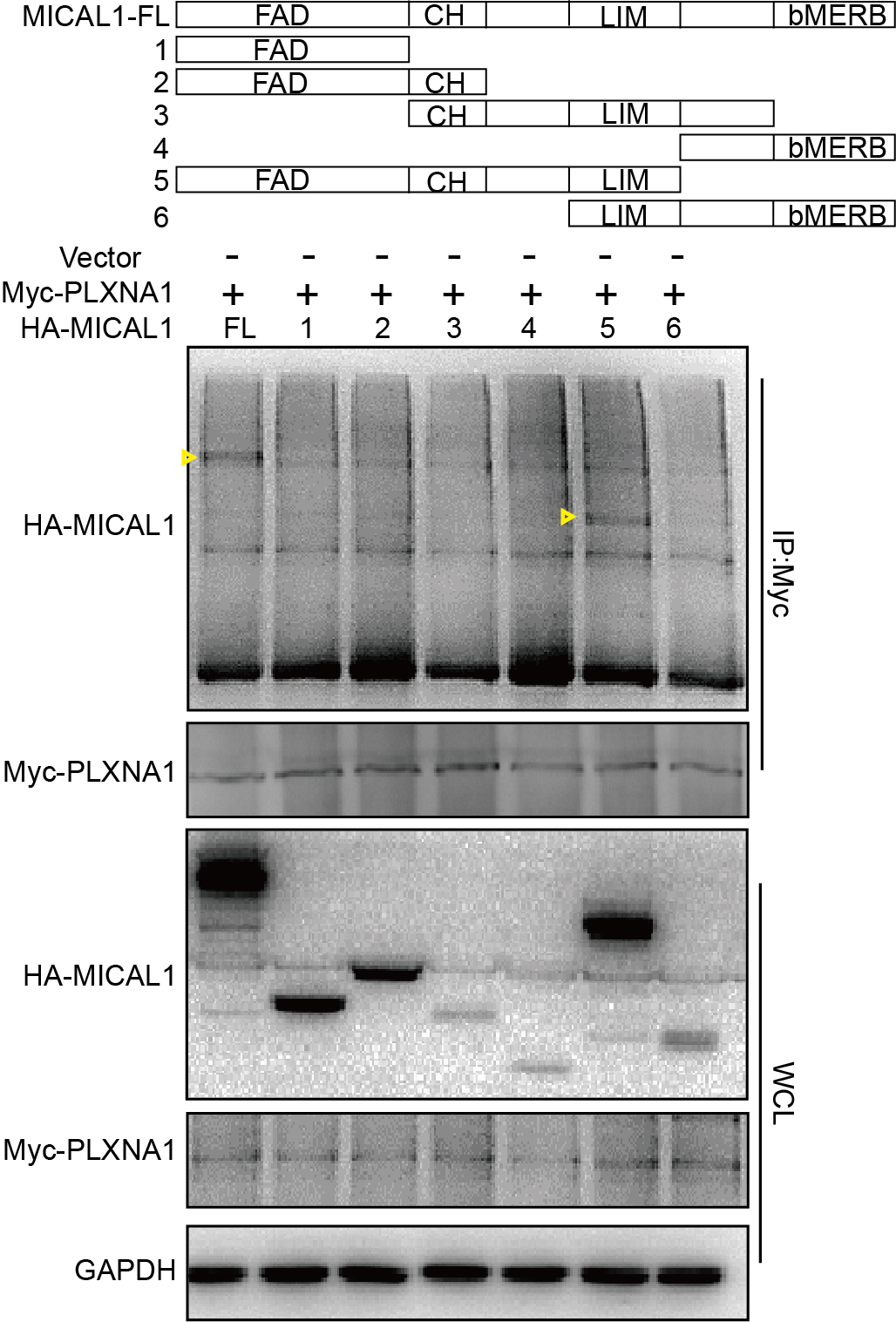
MICAL1 consists of N-terminal FAD domain, followed by CH, LIM, and C-terminal CC domains. Normally, MICAL1 is autoinhibited by its intramolecular interactions between the CC domain and the CH and LIM domains. Physical interactions between MICAL1 and proteins such as plexins and PAK1 may spatiotemporally relieve MICAL1 autoinhibition(Schmidt et al., 2008). Here, PlexinA1 overexpression was seen in gastric cancers. Silencing of PlexinA1 prevents MICAL1 expression and gastric cancer cell migration. Moreover, we observed that PlexinA1 interacted with MICAL1 in gastric cancer cells. Differently from the suggestion that the cytoplasmic portion of plexins bind to the CC domain of MICALs(Rajan et al., 2023), we found that PlexinA1 binds to the FAD + CH + LIM domains, but not the CC domain of MICAL1. Previous studies have shown that protein degradation can be regulated by two possible mechanisms: the proteasome-based or lysosomal-based proteolytic pathway. Our results demonstrate that only proteasome inhibitors could prevent the reduction in MICAL1 degradation by PlexinA1 knockdown. In addition, MICAL1 ubiquitination levels were increased in gastric cancer cells transfected with siPlexinA1. Together, these results indicate that the knockdown of PlexinA1 promotes the ubiquitination, and consequently proteasome-based degradation of MICAL1.
It has been found that the intracellular domain of plexins binds to the small GTPase Rac1(Zhang and Buck, 2017). This study was designed to explore whether PlexinA1 could also regulate Rac1 activation. We noticed that knockdown of PlexinA1 apparently suppressed Rac1 activation. Conversely, the effects were reversed by PlexinA1 overexpression, implying that PlexinA1 maintains Rac1 activation. When we further analyzed the mechanism about how PlexinA1 maintain of MICAL1 stability, we observed that activating Rac1 significantly abrogated the downregulation of MICAL1 in PlexinA1-depleted cells. Meanwhile, inhibiting Rac1 exerted the opposite effect, suggesting that PlexinA1 maintains MICAL1 stability through Rac1 activation. A recent study showed that Rac1 directly activates NADPH oxidases to produce ROS, which have been implicated in modulating breast cancer cell migratory properties(Tobar et al., 2008). ROS also account for cancer cell genomic instability, proliferation, angiogenesis, and resistance to apoptosis(Morry et al., 2017). Here, silencing of PlexinA1 significantly decreased ROS production, and ROS scavenger NAC suppressed the high levels of MICAL1 induced by the overexpression of PlexinA1 or increased activation of Rac1. Thus, it is likely that PlexinA1 maintains MICAL1 stability in a Rac1/ROS-dependent manner. Combined with the role of ROS in activating PAK1 phosphorylation(Yang et al., 2011), and phosphorylated PAK1 involves in relieving MICAL autoinhibition by phosphorylating serine 817 and 960 residues within the C-terminus of MICALs(McGarry et al., 2022), it can be suggested that Rac1 acts downstream of PlexinA1 to regulate MICAL1 stability and normal functions via ROS-dependent PAK1 activation. The mechanisms by which PlexinA1 regulates MICAL1 stability remain to be elucidated in detail.
It is widely known that vimentin expressions are associated with EMT, which is known to enhance cell migration and invasion(Nowak and Bednarek, 2021; Usman et al., 2021). Here, we found that knockdown of PlexinA1 and MICAL1 induced a significant reduction in vimentin expression as well as a impaired cell migratory ability; in contrast, both PlexinA1 and MICAL1 overexpression activated vimentin expression, consequently promoting cell migratory ability. MICAL1 has been reported to associate with vimentin through its C-terminal region(Suzuki et al., 2002), which implies an intimate connection between MICAL1 and vimentin. As an intermediate filament protein, vimentin is well-known to play a pivotal role during cell migration, including forming filaments to modulate focal adhesion, generating directional migratory force, and modulating genes for EMT inducers(Usman et al., 2021). Our study is the first, to our knowledge, demonstrated that PlexinA1 and MICAL1 contributes to gastric cancer cell migration, at least partially through maintaining vimentin expression.
In conclusion, PlexinA1 and MICAL1 regulates gastric cancer cell migration by inducing vimentin expression. PlexinA1 binds to and stabilizes MICAL1 expression in a Rac1/ROS-dependent manner, avoiding ubiquitination and following degradation of MICAL1. Although our results indicate that PlexinA1 and MICAL1 may serve as new therapeutic targets in gastric cancer field due to its potential to restrain tumor migration, their clinical evaluation needs to be investigated in the future.
{kind=link}
{kind=link}