Cells and drug treatments
We kept at 37 ˚C, two human TALL cell lines: Jurkat and CCRFCEM (obtained from the American Type Culture Collection; ATCC), in RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented with the following: 10% heatinactivated fetal bovine serum (FBS; HyClone; GE Healthcare Life Sciences), 1 mM sodium pyruvate (HyClone; GE Healthcare Life Sciences), 100 U/ml penicillin, and 100 µg/ml streptomycin (Invitrogen; Thermo Fisher Scientific, Inc.). Cells were maintained in a humidified atmosphere containing 95% air and 5% CO2. Cells were then treated with niclosamide (ACROS Organics™) at different doses and time courses.
Primary T-ALL cells
Primary T-ALL cells were harvested from PBMCs of either patients with T-ALL or healthy controls in Taichung Veterans General Hospital. For PBMC culture, 8 mL of blood were collected in sodium citrate tubes (Vacutainer® CPT™, BD, USA) after being taken from patients or healthy controls. PBMCs were purified through centrifugation down a density gradient. Cells were cultured in RPMI-1640, supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, 25 mM HEPES, and 2 mM L-glutamine. The study protocol was approved by the Institutional Review Board of Taichung Veterans General Hospital, Taiwan (NO. CG19384A).
Isolation of RNA
Cells were collected from in vitro treatments, and harvested after drug treatments. Total RNA was extracted using the Amersham RNAspin Mini Kit (Cytiva) according to manufacturer's instructions.
RNA sequencing
The quality of the raw reads were checked using FastQC (v0.11.9). Adapters, low quality bases and reads were trimmed with Trimmomatic (v0.36), using these parameters ILLUMINACLIP: Adapter.fa:2:30:10 LEADING: 3 TRAILING: 3 SLIDINGWINDOW: 4:15 MINLEN: 36. After trimming, clean reads were aligned to the reference genome using HISAT2 (v2.2.1). StringTie (2.2.1) was used to normalize read counts. FeatureCounts (v1.6.2) were used to summarize reads. Differential expression analyses were performed using either DEGseq (v2.2.1) without biological replicates, or DESeq2 (1.34.0) with biological replicates. Differential expressions of gene sets were filtered based on the absoluteon value of log2fold change ≥ 2 and adjusted p-value < 0.005 according to results of DEGseq. Differentially expressed gene sets at statistical significance were filtered based on the absolute value of log2fold change ≥ 2 and adjusted p-value < 0.05 according to the results of DESeq2. To identify biological processes and pathways that are significantly enriched by the differentially expressed genes, the gene set enrichment analyses (or GSEA) were performed with the ClusterProfiler (4.2.2). The database of the Kyoto encyclopedia of genes and genomes (or KEGG) was used for annotation. Pathway visualization was performed using Pathview (1.34.0).
CCK-8 assay
Jurkat and CCRFCEM cells were seeded at a density of 1x105 cells/well in a 96well cell culture plate. Jurkat cells were treated at 37˚C for 24 h, with either vehicle control (DMSO) or with different doses of niclosamide. Afterward, cell viability was determined with the CCK-8 kit (cat. no.96992, Sigma-Aldrich) according to the manufacturer's instructions. In addition, optical density was measured at 450 nm with a microplate reader (BioTek Synergy HT).
Western blot analysis
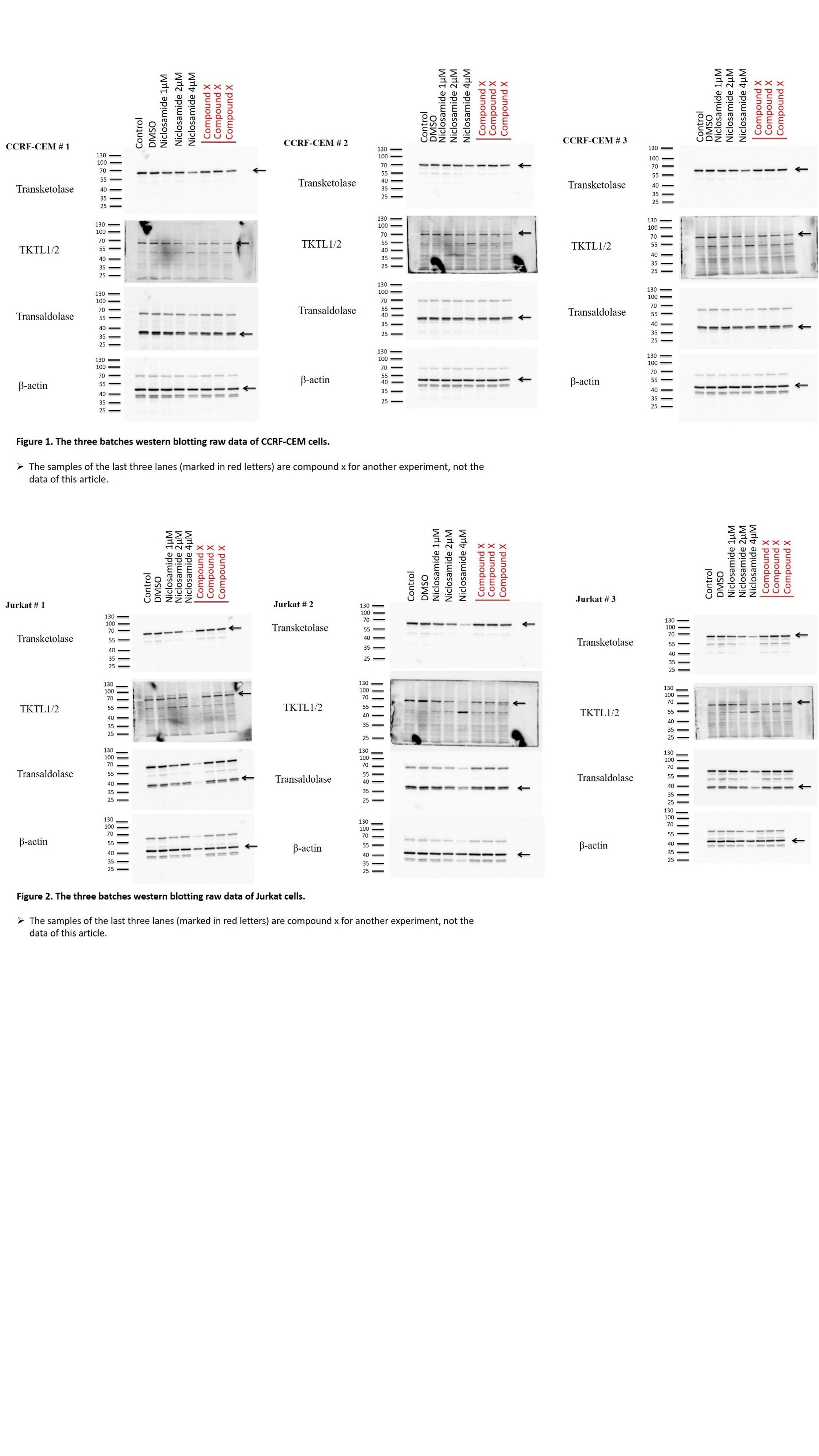
Jurkat and CCRFCEM cells were seeded at a density of 1.5 x106 cells/well in a 12well cell culture plate. Jurkat cells were treated with either vehicle control (DMSO) or niclosamide (1.0, 2.0 and 4.0 µM). CCRFCEM cells were treated with either vehicle control (DMSO) or niclosamide (1.0, 2.0 and 4.0 µM). Both cell cultures were kept similarly at 37 ˚C for 24 h. After niclosamide treatment, cells were harvested and levels of protein expressions were evaluated with western blotting using appropriate antibodies. In brief, cells were first washed with PBS before collection. Cells were lysed in RIPA buffer (Biomed, Taiwan), and centrifuged at 13,000 g for 10 min at 4 ˚C. The supernatant was used for protein quantification with a Coomassie protein assay reagent (Thermo Fisher Scientific, Inc.). Fixed amounts of protein (15 µg/lane) were loaded into each well and separated by SDSPAGE. Samples were finally transferred to the PVDF membrane. Immunoblotting was conducted using the following antibodies: Transketolase (TKT) (cat. no. sc-390179; Santa Cruz; 1:1,000 dilution), Transketolase Like 1/2 (TKTL1/2) (cat. no. sc-514513; Santa Cruz; 1:500 dilution), Transaldolase (cat. no. sc-166230; Santa Cruz; 1:1,000 dilution) and βactin (cat. no. 4970S; Cell Signaling; 1:5,000 dilution), followed by incubation with secondary antimouse IgG, HRPlinked antibody (cat. no. 7076P2; Cell Signaling Technology; 1:7,000 dilution) or goat antirabbit IgG antibody (cat. no. 7074P2; Cell Signaling Technology; 1:7,000 dilution). Labeled proteins were detected using the ECL Detection Kit (Millipore) from the Alliance Q9 (UVITEC, UK). Normalization was performed with the βactin antibody.
Measuring ATP levels
ATP measurements were carried out with the ATP Assay Kit Colorimetric/Fluorometric kit (ab83355, Abcam, USA) according to the manufacturer’s instructions. In brief, 1 x 106 cells were washed in cold PBS and then homogenized in 100 µl of ATP assay buffer. Cells were centrifuged at 4 oC at 13,000 g and then the supernatant was collected. Standard samples were prepared according to manufacturer instructions. Test samples and background controls were prepared in a 96-well plate according to manufacturer’s instructions for colorimetric assay. Finally, plates were read with a plate reader (Enspire 2300-0000/PerkinElmer) at OD 570 nm.
TKT activity assay
TKT activity was measured with the Transketolase Activity Assay Kit (Fluorometric) (ab273310, Abcam, USA) according to the manufacturer’s instructions. In brief, cells (4 x 105 ) were homogenized in a 100 µl TKT Assay buffer for lysis, and then centrifuged at 4 ° C at 10,000 g. Supernatants were then collected. Standard samples were prepared according to manufacturer instructions. Test samples and background control were prepared in a 96-well plate according to manufacturer instructions for fluorometric assay. Fluorescence was measured immediately at 30 sec intervals for 30 to 45 min at 37°C.
Seahorse analyses
Cells were plated in Seahorse XF 6-well plates at respective optimal densities one day prior to measurements. These cells were subjected to the experimental conditions as described above, and incubated in Seahorse XF Assay Media at 37°C for 1 hr without CO2 just before assay. Substrate concentrations were as follows: 1 µM for Oligo and FCCP, 1 µM /0.5 µM for Rot/AA, and 5 mM for succinate. All reagents were from Seahorse Bioscience. Basic conditions of glycolysis, maximal glycolytic capacity, and non-glycolytic activity were measured with the Seahorse XF-6 Extracellular Flux Analyzer.
T-ALL xenograft murine model
Female NOD/SCID mice aged 8 weeks with body weight 18 to 22 g were obtained from the National Laboratory Animal Center (Taipei, Taiwan). Mice were housed under specific pathogen-free conditions with 12:12-h dark/light cycle, with food and water ad libitum. All experiments were approved by the Animal Care and Use Committee of Kaohsiung Veterans General Hospital (IACUC NO. 2021-2022-A003-MOST). CCRFCEM cells (1x106 cells/100 µl/mouse), suspended in a 1:1 mixture of BD Matrigel™ (basement membrane matrix, growth factor reduced, phenol redfree; BD Biosciences, cat. no. 356231) and RPMI-1640 medium were injected subcutaneously into the flank region of NOD/SCID mice. Tumors were periodically measured with a digital caliper 3 times/week until sacrifice. Mice were randomly divided into different experiment groups (6 mice/group) that included: (a) vehicle control group, (b) niclosamide (20 mg/kg) treatment group, (c) α-KG (10 mg/kg) treatment group, and (d) α-KG (10 mg/kg) plus niclosamide (20 mg/kg) treatment group. Niclosamide and α-KG treatments were delivered through intraperitoneal injections, 3 times/week, beginning on day 7 after induction. Mice were sacrificed on day 28, and tumor samples collected for H&E and IHC stainings.
Hematoxylin and eosin (H&E) staining
Tumor tissues were removed from the xenograft mice and fixed in 10% formalin for 24 h at room temperature. Tissues were embedded in paraffin, and then sectioned for staining with hematoxylin and eosin (H&E). Microscopic images were captured under light microscopy (AXIOVERT, ZEISS).
Immunohistochemistry (IHC) staining
After tumor tissues had been removed from the xenograft mice, specimens were fixed in 10% phosphatebuffered formalin, dissected and then embedded in paraffin. Paraffin sections (3 µm in thickness) were incubated with 0. % hydrogen peroxide for 15 min, blocked for 1 h at room temperature, and then incubated with Transketolase (TKT) (cat. no. sc-390179; Santa Cruz) and Ki-67 (cat. no. 9449S; Cell Signaling) overnight at 4 ˚C. After washing with TBST, sections were processed with the Epredia™ UltraVision™ Quanto Detection System HRP DAB kit (Epredia™ TL-060-QHD), and when required, immediately stained with DAB (Epredia™).
Statistical analyses
Data were expressed as mean ± standard error of mean. Statistical analyses were performed using oneway ANOVA followed by Tukey's test for posthoc comparisons. Results were analyzed using the software GraphPad Prism (version 6; GraphPad Software, Inc.). Statistical significance was set at p < 0.05.
{kind=link}