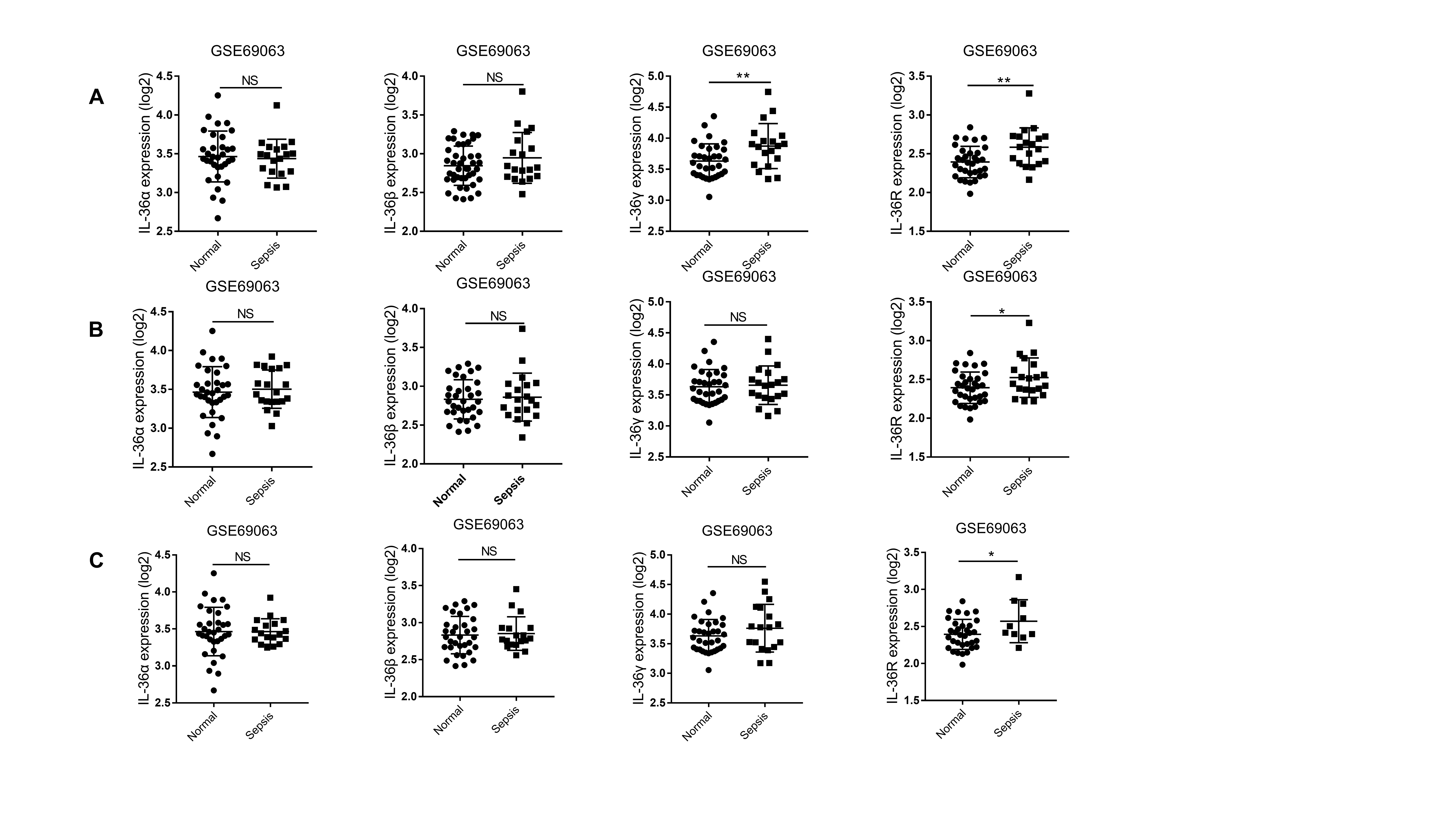
The clinical role of IL-36 in diagnosing sepsis and predicting 28-day mortality
Higher serum IL-36 subtypes levels were related to lower SOFA scores in the patients with sepsis on the day of admission (Fig. 2A). Moreover, there was a significant correlation between IL-36 subtypes and PCT (Fig. 2B), or CRP (Fig. 2C) levels on admission.
The ROC curve of IL-36 subtypes for diagnosing sepsis is shown in Fig. 3. The AUC of IL-36α on the day of admission was 0.797 (p < 0.001, [95% CI] 0.693–0.797). The AUC of IL-36β on the day of admission was 0.681 (p = 0.005, [95% CI] 0.555–0.681). The AUC of IL-36γ on the day of admission was 0.863 (p < 0.001, [95% CI] 0.776–0.863).
In addition, we analyzed whether each IL-36 isoform contributes to predicting 28 days in septic patients. For the prediction of 28-day mortality (Fig. 4), the area under the ROC curve for IL-36α on the day of admission was 0.730 (p = 0.004, [95% CI] 0.572–0.730), higher than the AUC for IL-36β (AUC = 0.706; [95% CI] 0.547–0.706, p = 0.011) but lower than the AUC for IL-36γ (AUC = 0.756; [95% CI] 0.589–0.756, p = 0.003) and SOFA score (AUC = 0.738; [95% CI] 0.578–0.897, p = 0.011).
IL-36 receptor deletion aggravated CLP-induced sepsis mortality
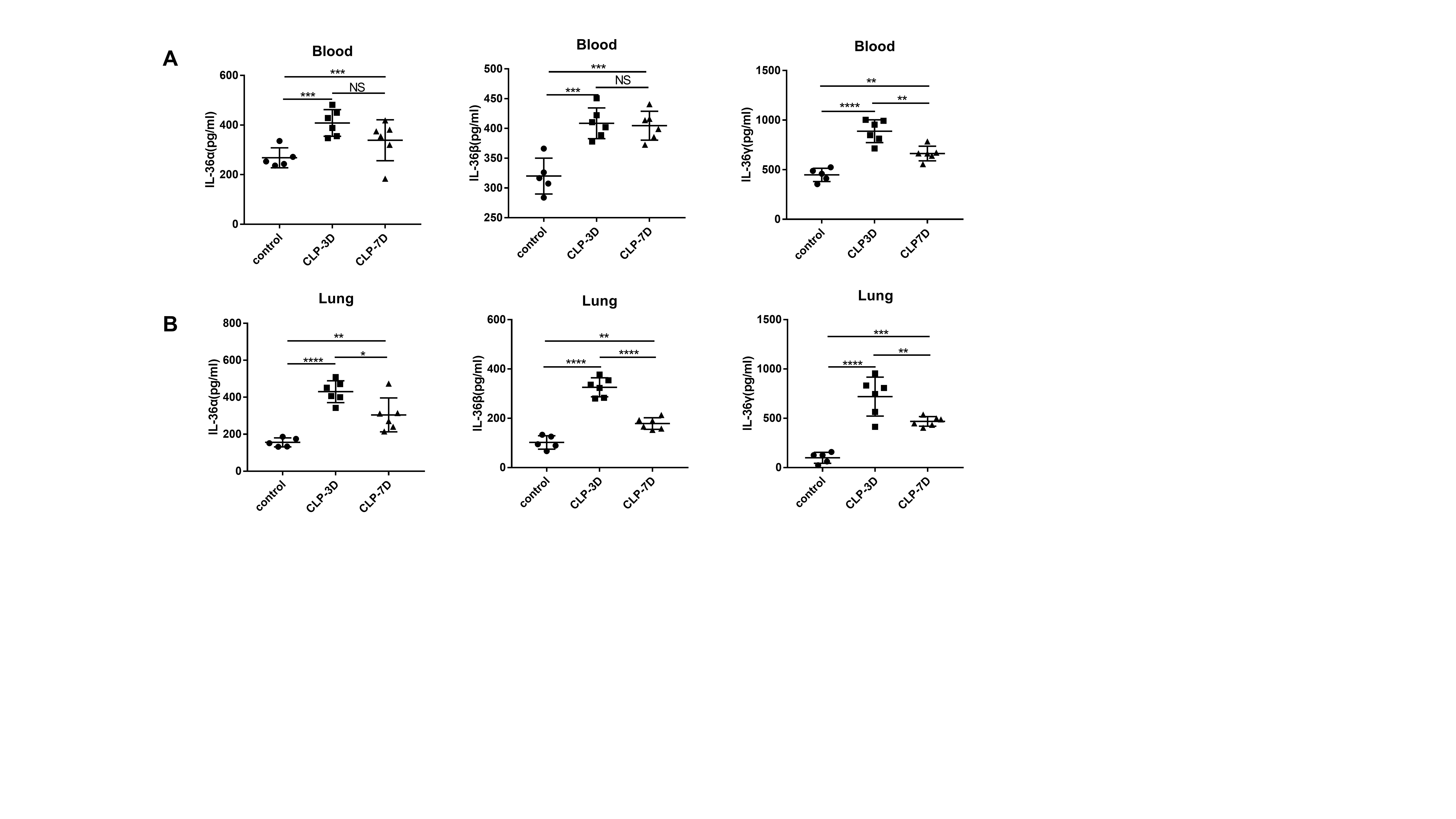
To investigate whether levels of IL-36 family members are elevated in septic mice, we generated a sepsis model and examined protein expression levels of IL-36α, IL-36β, and IL-36γ during sepsis. Both IL-36α, IL-36β, and IL-36γ levels were significantly elevated in serum and lungs at 3-day and 7-day post-CLP challenge(Fig. S2). The above data suggest that IL-36 family member's production is also significantly increased during mouse sepsis, indicating that IL-36 signaling may be involved in the pathogenesis of sepsis.
IL-36α, IL-36β, and IL-36γ all bind only to the IL-36 receptor (IL-36R). To examine whether IL-36R mediated signaling is involved in the host response against CLP-induced polymicrobial sepsis, WT and IL-36R−/− survival were assessed out to 7 days. IL-36R deletion dramatically decreased septic mouse survival compared with the WT septic mice (Fig. 5A).
Exacerbated mortality of IL-36R−/−septic mice was closely associated with increased organ injury. Serum concentrations of ALT and AST, markers for hepatocellular injury, and creatinine, a marker for renal failure, were significantly increased in IL-36R−/− septic mice at 3 days after CLP (Fig. 5B).
To determine whether there is any difference in bacterial clearance, the homogenate diluents of lungs, livers, kidneys, hearts, and spleens were cultured on blood agar plates. Vital organs bacterial CFU levels were significantly increased in the IL-36R−/−mice compared to WT mice (Fig. 5C). And CFU levels in the lung were higher than that in other organs (Fig. 5C).
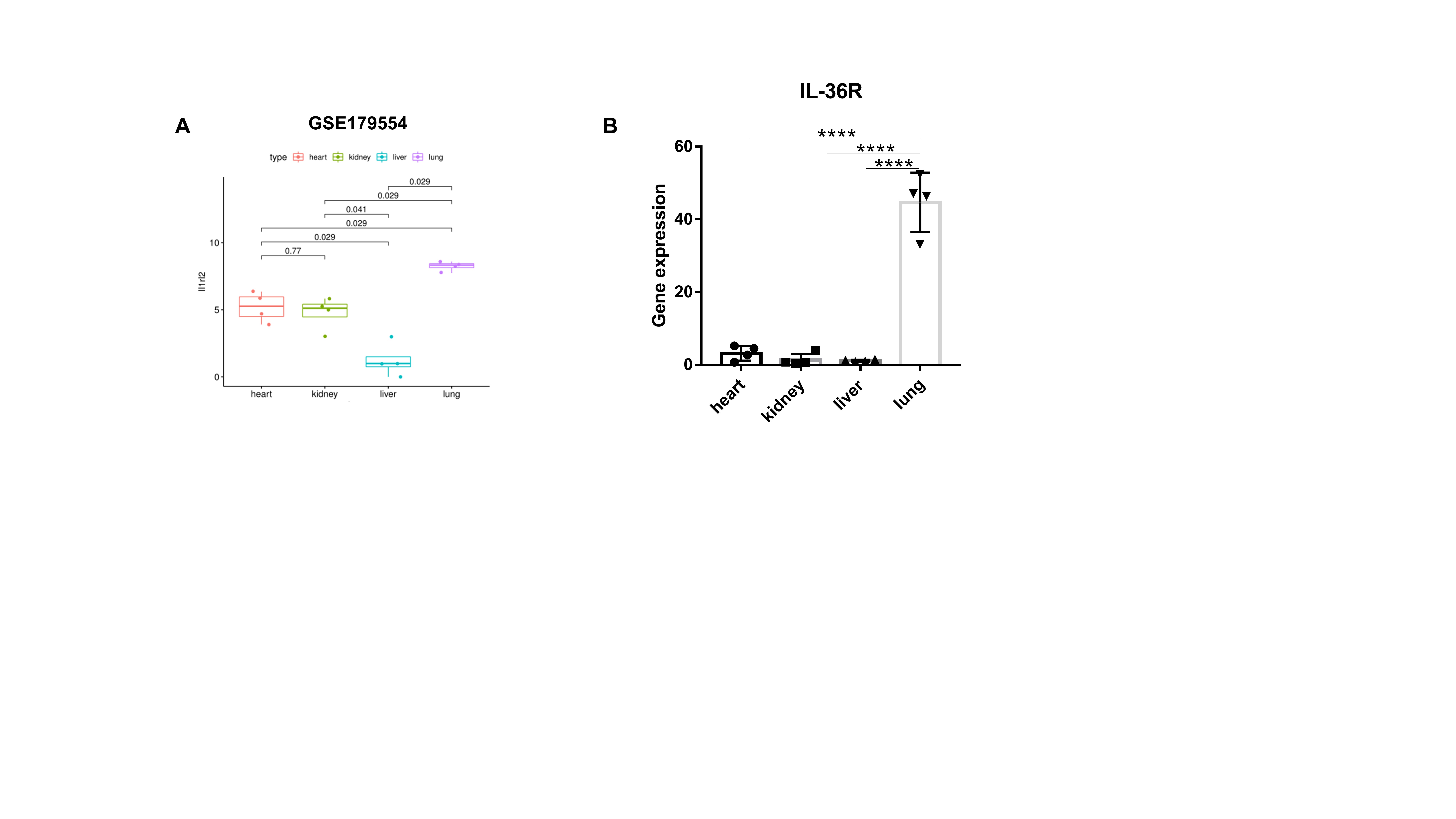
IL-36R is predominantly expressed in the lungs of mice by analyzing a published RNA-Seq dataset (GSE179554) (Fig. S3A). The expression of IL-36R ligands was also analysed by quantitative PCR in heart, kidney, liver, and lung (Fig. S3B).
In addition, IL-36R knockdown significantly increased pathological scores in lungs (Fig. 6A,B,C,D,E,F and G) 3 days after CLP surgery.
Neutrophil recruitment to the inflamed lung was increased in IL-36R−/− mice, as analyzed by FACS (Fig. 6H and I).
IL-36R Deficiency in Nonhematopoietic Cells Served as the Major Contributor to the Exacerbation of Sepsis.
To assess the relative contribution of hematopoietic and nonhematopoietic cells to the sepsis exacerbation in IL-36R−/−mice, we used reciprocal bone marrow (BM) transplantation and generated the following chimeric mice: WT → WT, IL-36R−/−→ WT, WT → IL-36R−/−, and IL-36R−/−→ IL-36R−/− (Fig. 7A). At 8 week after BM transplantation, mice were subjected to CLP. During a 7-day experimental period, we found that mice with IL-36R deficiency in the nonhematopoietic compartment (WT → IL-36R −/−and IL-36R −/−→IL-36R −/−) suffered from significantly higher mortality rates (Fig. 7B). Serum concentrations of ALT and AST, markers for hepatocellular injury, and creatinine, a marker for renal failure, were significantly increased among IL-36R−/− deficiency in the nonhematopoietic compartment mice at 3 days after CLP (Fig. 7C). Lung bacterial CFU levels were significantly increased in the mice with IL-36R deficiency in the nonhematopoietic compartment (WT → IL-36R −/−and IL-36R −/−→IL-36R −/−) (Fig. 7D). Neutrophils were also significantly enhanced in lung tissues resulting from IL-36R deficiency in nonhematopoietic cells (WT → IL-36R −/−and IL-36R −/−→ IL-36R −/−) at 3 d after CLP (Fig. 7E and F). Histologically, mice with IL-36R −/−nonhematopoietic cells (WT → IL-36R −/−and IL-36R −/−→ IL-36R −/−) displayed extensive destruction of lung architecture and higher injury scores in response to CLP (Fig. 7G and H). Thus, these data suggested that IL-36R deficiency in nonhematopoietic cells, but not in BM-derived cells, exacerbated sepsis lethality and lung injury processes.
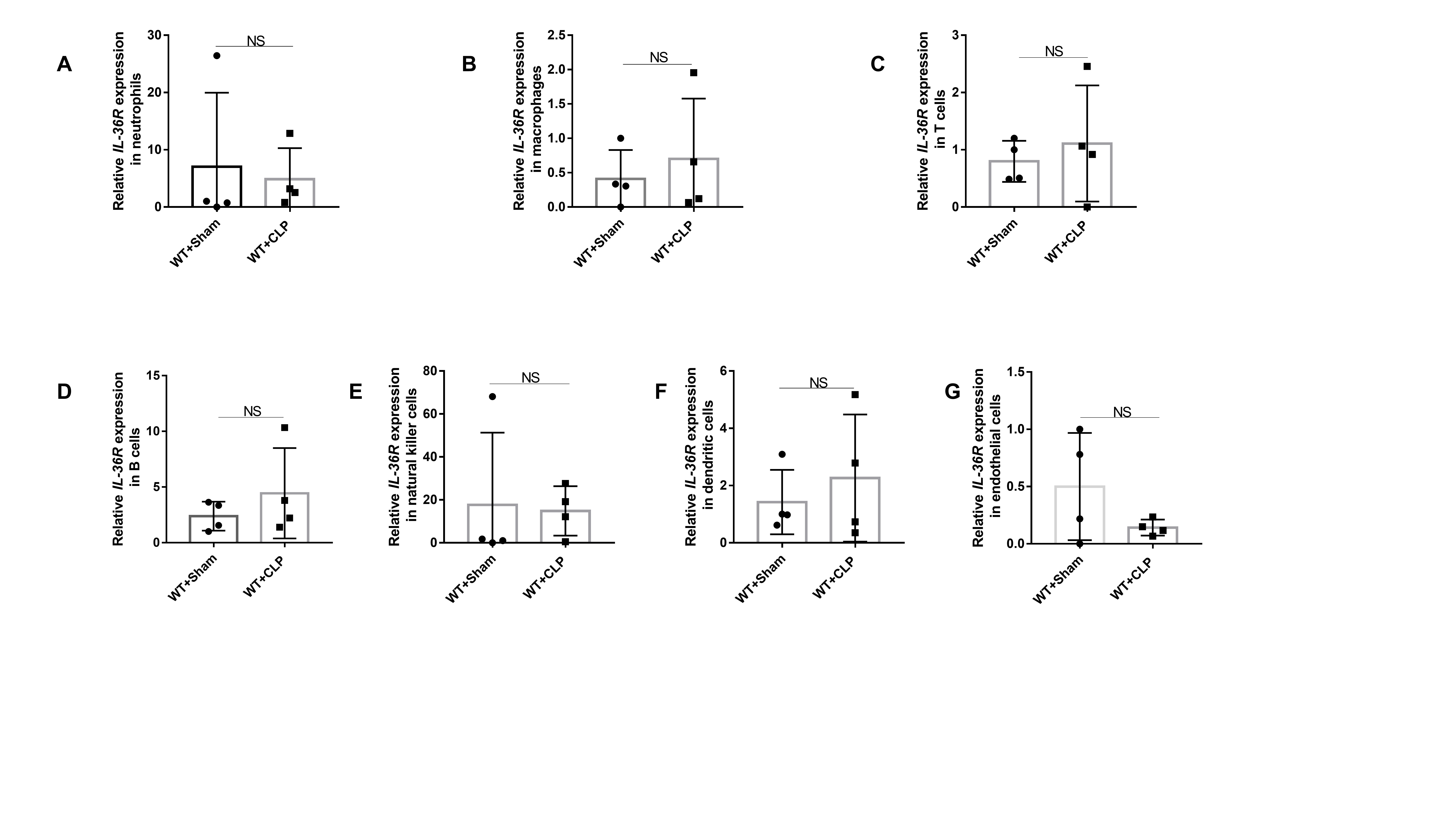
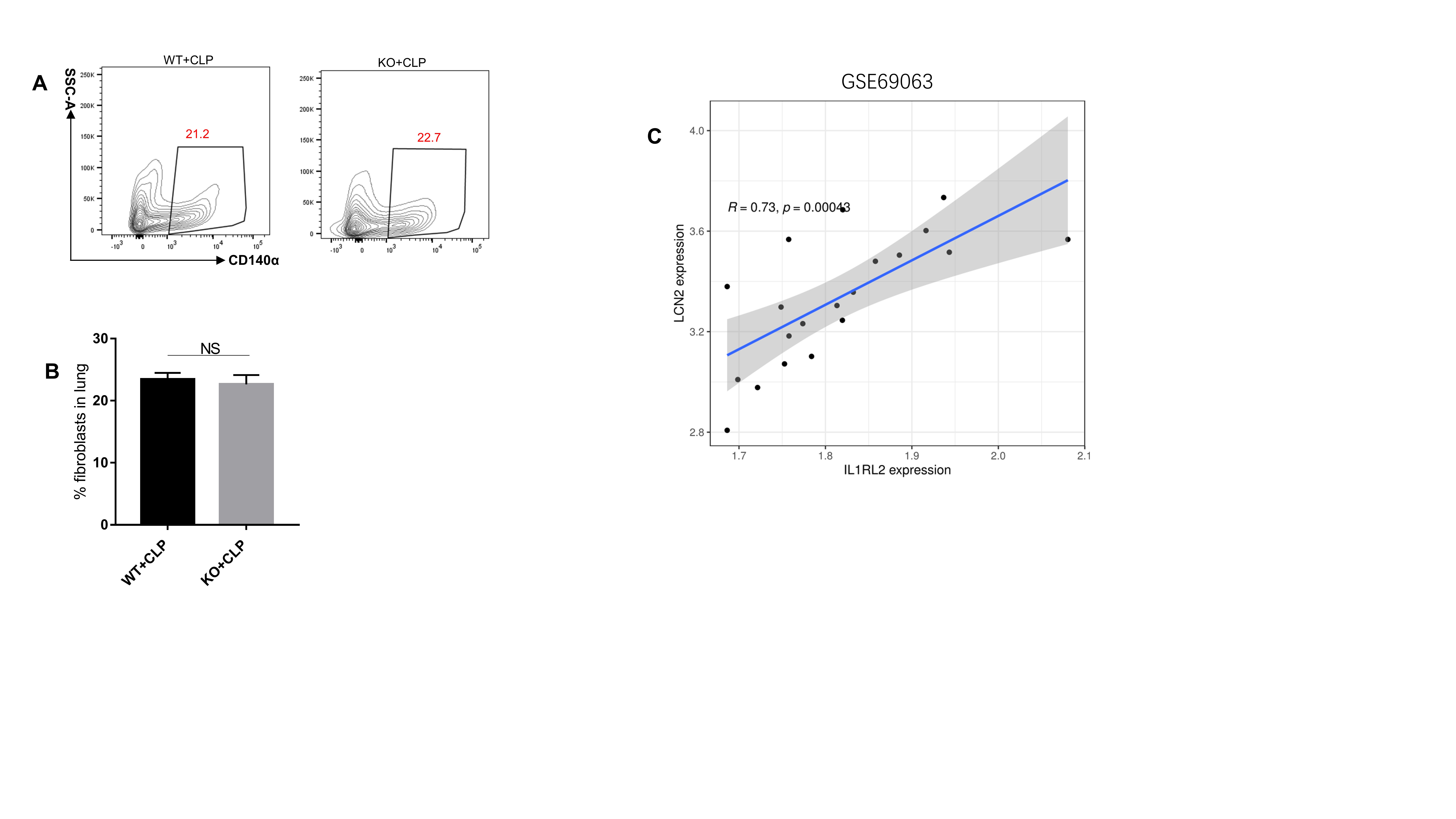
To identify target cell types deficient in IL-36R, we first assessed the type of each lung resident cell following sepsis, particularly the level of IL-36R expression in non-hematopoietic cells. By conducting qRT-PCR analysis for sorted lung cells, we definitely did not find any increase in IL-36R expression in neutrophils, macrophages, dendritic cells, or other hematopoietic cells after sepsis(Fig. S4A-F). There is also no difference in IL-36R expression on endothelial cells (Fig. S4G). We found that IL-36R expression was significantly elevated on fibroblasts and epithelial cells following sepsis, which are considered critical non-hematopoietic cells involved in the pathogenesis of septic lung injury(Fig. 8A and B). We could also observed that IL-36R is strongly expressed in lung fibroblasts and epithelial cells with sepsis as detected by immunofluorescence(Fig. 8C and D). The proportion of lung fibroblasts between KO and WT mice were not different after sepsis (Fig. S5A and B). By mining a recently published lung single-cell RNA sequencing dataset on CLP-induced sepsis (GSE207651), we found that antimicrobial protein lipocalin2 (LCN2) expression was significantly elevated on fibroblasts following sepsis (Table S4). Moreover, there was a significant correlation between IL-36R and LCN2 according to the published databases (Fig. S5C). After sepsis, LCN2 was predominantly decreased in lung fibroblasts among KO mice (Fig. 8E) and the same results as detected by immunofluorescence (Fig. 8F).
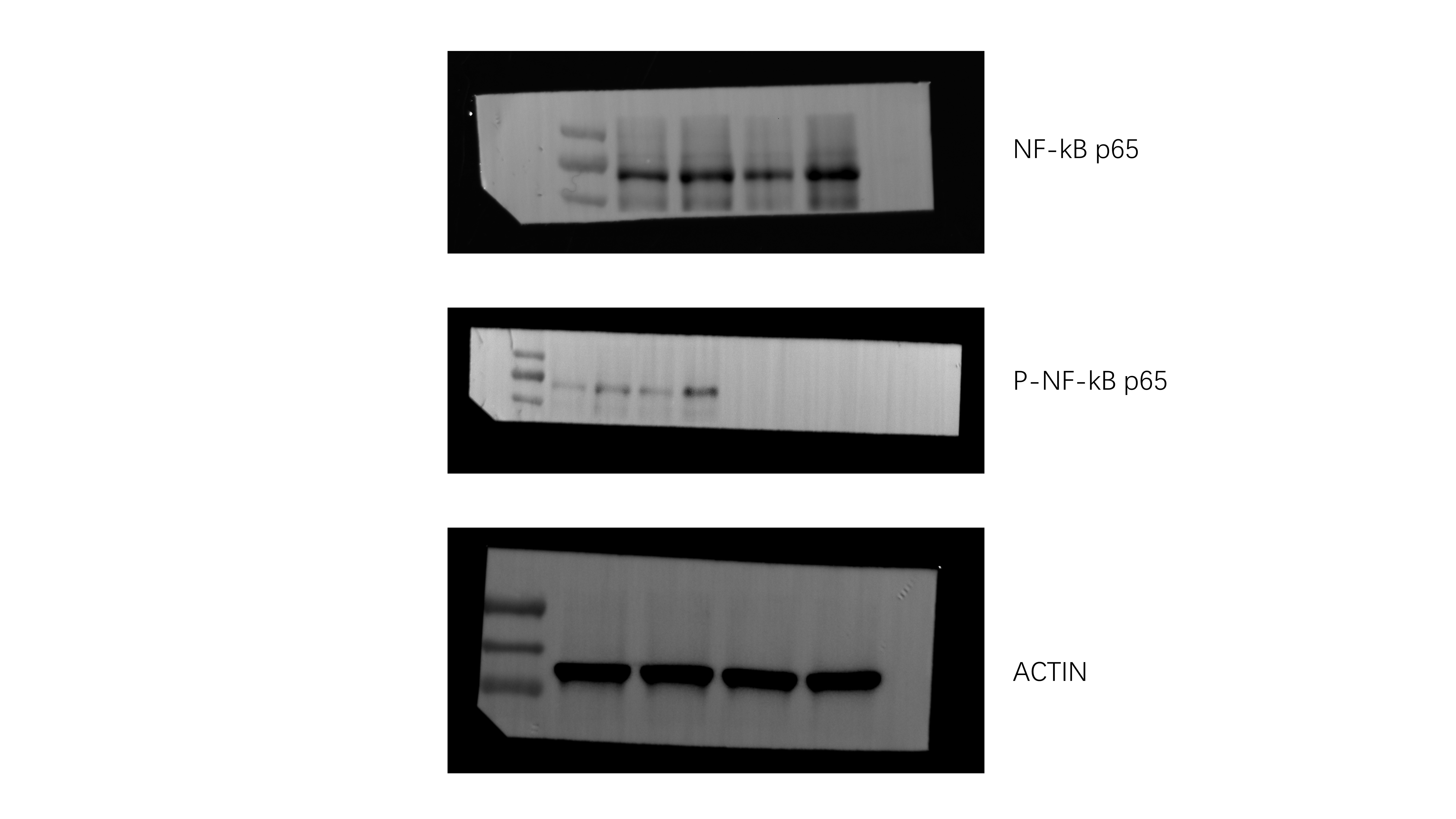
To assess the role of epithelial cell in this process, we also examined the propotion of lung epithelial cells between KO and WT mice. We found that the proportion of lung epithelial cells significantly decreased among KO mice after sepsis (Fig. 9A). To determine whether IL-36R deletion could functionally influence the lung epithelial cells death, we analysed the death of epithelial cells by 7-AAD and Annexin V. We detected a marked induction of epithelial cells that stained positive for Annexin V and negative for 7-AAD. These results indicating that IL-36R deletion could promote the early apoptosis of the lung epithelial cells (Fig. 9B). In subsequent studies, we wanted to perform a comprehensive analysis of the molecular mechanisms induced by IL-36R signalling in lung epithelial cells. Therefore, we sorted epithelial cells from the lung of WT and KO mice in the presence CLP for 3 days. Subsequently, RNA was purified from both groups and whole genome expression profiling was performed by RNA-seq. RNA-seq data have been deposited in the public database GEO with accession number GSE240924. Hierarchical clustering of samples showed high similarities in overall transcription patterns upon IL-36R knockout(Fig. 9C). Gene ontology (GO) based functional annotation analysis with the differentially expressed and upregulated genes revealed that there was a significant enrichment for early apoptosis in lung epithelial cells with IL-36R deletion (Fig. 9D). Through KEGG analysis, the nuclear factor κB (NF-κB) pathway on lung epithelial cells was activated with IL-36R deletion (Fig. 9E). In summary, the RNA-seq studies with primary lung epithelial cells demonstrated that such cells are broadly modulated by IL-36R activation resulting in characteristic gene expression patterns with NF-κB. The results of WB are consistent with those of RNA-seq (Fig. 9F).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}