Isolation and culture of hUC-MSCs
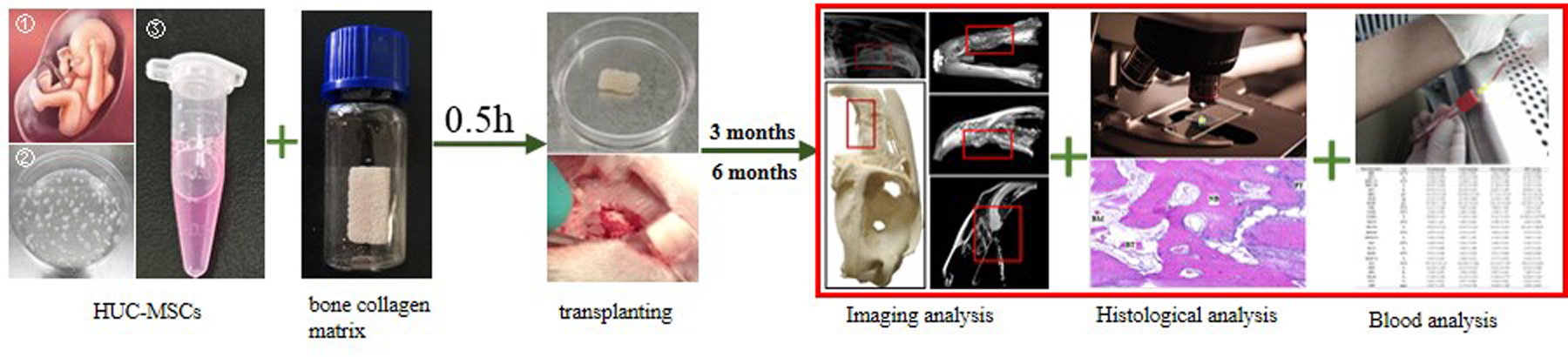
The hUC-MSCs were obtained by tissue mass culture. Briefly, the blood vessels of the human umbilical cord were removed, and then the cord was cut into approximately 1 mm3 of tissue. Until tissue block was attached to the bottom of the cell culture dish, α-Minimal Essential Medium (α-MEM) culture medium containing 10% fetal bovine serum, 100 IU/mL penicillin, and 10 mg/mL streptomycin was added to the dishes and cultured in a carbon dioxide incubator. The cell growth was monitored, and the tissue was removed when the cells had radially covered the surface of the culture plate. The surface antigens of within 5 passages cells were detected using a human MSC assay kit (BD Biosciences Franklin Lakes, NJ, USA).
Preparation Of Implant Materials
The bone collagen matrix is a heterogeneous bone matrix prepared from bovine cancellous bone refined in a series of processes, thereby retaining its natural three-dimensional porous structure. The main components of the bone collagen matrix are hydroxyapatite and collagen. The collagen membrane coverings the site after the bone collagen matrix was implanted. The collagen membrane was approximately 0.8 mm thick. Before use, the collagen membrane was cut into small pieces the same size as the defect area. The bone collagen matrix and collagen membrane were both provided by Zhenghai Bio-technology Co., Ltd (Yantai, China). The hUC-MSCs within five passages were harvested and suspended in phosphate buffer solution (PBS) at the concentration of 107 cells/ mL.
Groups And Treatment
In this study, 48 female JWRs (bodyweight: 2000 ± 300 g, about two-month-old) were used. The JWRs were purchased from Huafukang Bio-technology Co., Ltd (Beijing, China). All these animals were kept in the animal room at the National Research Institute for Family Planning. and were provided with clean water and fresh food. The indoor conditions were as follows: temperature (24 ± 1℃); air humidity (55%±5%); noise (less than 60dB); lighting time (12 h). The room always was kept clean, dry and ventilated. The experimental design and implementation were approved by the local research and ethics committee.
In this study, JWRs were randomly assigned to each of four groups: normal, control, material, and MSCs. The rabbits were first anesthetised by an intravenous injection of serazine hydrochloride into the ear margin (concentration: 1–2 mg/kg) and then the left maxilla was located. The alveolar process cleft model was established by removing part of the jawbone equal to the volume of 1cm ⋅ 0.5 cm ⋅ 0.4 cm with a rongeur (Fig. 1).
The normal group was fed normally. In the control group, after part of the jawbone was removed, the collagen membrane was used to directly cover the injury, and the muscles and skin at the injured site were sutured. In the material group, part of the jawbone was removed, the same volume of the bone collagen matrix was implanted in the injury, and then the collagen membrane was used to directly cover the injury, and the muscles and skin of the injured site were sutured. In the MSCs group, bone collagen matrix (1cm ⋅ 0.5 cm ⋅ 0.4 cm) was inoculated with 107 cells/mL hUC-MSCs suspension and cultured in a carbon dioxide incubator for 0.5 h. After part of the jawbone was removed, combining bone collagen matrix with hUC-MSCs was implanted and then the collagen membrane was used to directly cover the injury, and the muscles and skin of the injured site were sutured. Conventional anti-inflammatory therapy (i.e. penicillin potassium: 4000 IU/kg/d) was then given to all the rabbits for 1 week to prevent postoperative infection. The rabbits were removed from each group at 3 and 6 months after the surgery. The rabbits were euthanised by intravenous injection of an overdose of serazine hydrochloride in the ear margin. The skull was removed, and partial skulls were placed in 4% NaOH and 95% ethanol respectively for 24h, and then taken out and placed in an oven to dry. Finally, the appearance of the skull tissue was photographed and marked. Partial fresh skull tissue was first fixed in 4% paraformaldehyde for 24h, de-calcified with 10% ethylenediaminetetraacetic acid (EDTA) for 1 month, and finally embedded in paraffin. The paraffin sections (6 µm) were prepared using a rotary microtome (Leica RM2245, Leica, GmbH, Germany). For histology staining, the paraffin sections were first de-paraffinized in xylene and then re-hydrated in graded alcohol solutions to pure water.
X-ray Analysis
X-ray analysis was performed using a SOFTEX® M-60 X-ray machine (Kanagawa, Japan) operated at 80 kV and 125 mA on the tissue samples prepared from the surgical sites in each group at 3 and 6 months after the surgery at the Beijing Ornamental Animal Hospital. The exposure time was 40 millisecond (ms).
Serum Bone Gla Protein (Bgp) Analysis
Three rabbits were randomly selected from each group at 3 and 6 months after the surgery. Approximately 3.5mL of blood was collected by ear-vein sampling. Some of the blood was used for direct detection and the remainder for serum separation. The blood routine, liver function, renal function and serum BGP of the rabbits were all measured. The routine blood tests were performed using an LH 750 automated haematology analyser (Beckman Coulter, USA). The blood biochemistry test was performed using a DXC 800 automated biochemical analyser (Beckman Coulter, USA).
Bone Formation Rate Analysis
Three rabbit skull models were randomly made in each group at 3 and 6 months after surgery, respectively. The lateral view of the surgical side of the rabbit was obtained by photographing. Then, the actual bone defect area and the total bone defect area were measured by Image J, and the difference value between the two was the area of new osteogenesis. The percentage of osteogenic area to defect area is the bone formation rate.
Micro-focus Computed Tomography (Micro-ct) Analysis
Three rabbits were randomly selected from each group at 3 and 6 months after the surgery. A skull model was prepared. The general appearance of the skull was recorded laterally and vertically. The bone re-generation in the material transfer area in the material transfer area were evaluated using a micro CT system (SIEMENS Inveon™ Research Workplace 4.2, Beijing). The repair of the maxillary region in each group was observed stereoscopically. In order to analyse the the quality of the newly formed bone in the defect area, three 1 mm3 areas were randomly selected from the centre of the cubic bone-defect area in each group to record the bone trabeculae and mineral density.
Hematoxylin And Eosin (He) Staining
HE staining was used to observe the tissue morphology. The nucleus and other areas were stained blue and red by the hematoxylin and eosin respectively. A TE2000-U inverted phase contrast microscope (Nikon, Tokyo, Japan) was used to observe the changes in the bone histomorphology.
Periodic Acid-schif (Pas) Staining
PAS staining was used to assess the glycogen concentration and was performed using a commercial kit (Senbeijia Biological Technology Co. Ltd, NanJing, Jiangsu, China) according to the manufacturer’s instructions. Briefly, the sections were incubated in the dark first with periodic acid solution and then with the Schiff reagent for 5 and then 20 min at room temperature, respectively. The sections were then counterstained with Lillie-Mayer's hematoxylin and were observed using differential interference contrast (DIC) optics microscopy (DM IL LED, Leica GmbH, Germany). The cartilage structure can be dyed either deep purple or red.
Sirius red staining
Sirius red staining was used to detect different collagen fibers and was performed using a commercial kit (Senbeijia Biological Technology Co. Ltd, Nanjing, Jiangsu, China) according to the manufacturer’s instructions. Briefly, the sections were incubated with sirius red for 1 h at room temperature and counter-stained with Lillie Mayer’s hematoxylin. Sirius red can dye type 1 collagen bright orange. Image J software was used to calculate the relative percentage of the type 1 collagen stained area under different fields in each group.
Bone-specific alkaline phosphatase (ALP) assay
Bone-specific ALP is an osteoblasts phenotypic markers, which can directly reflect the activity or function of osteoblasts. ALP calcium-cobalt staining was used to detect the bone-specific ALP content by a commercial kit (KeyGEN BioTECH Co.Ltd, NanJing, Jiangsu, China) used according to the manufacturer’s instructions. Briefly, the sections were incubated with ALP solution for 5 min and then with cobalt nitrate solution for 2 min at room temperature. The sections were then counter stained with eosin. The osteoblasts can be dyed black.
Immunohistochemical Staining For Bone Morphogenetic Protein 2 (Bmp-2)
After the sections were de-waxed, re-hydrated and subjected to heat-induced epitope retrieval, they were incubated with rabbit anti-BMP-2 polyclonal antibody (1:1000; Abcam, ab6285, Cambridge, UK), and then incubated with HRP-conjugated goat anti-rabbit IgG (1:5000; Jackson ImmunoResearch Laboratories, West Grove, Pennsylvania, USA). Normal rabbit serum was used as blocking solution. The sections were developed with diaminobenzidine tetrahydrochloride (DAB) and counterstained with hematoxylin. Samples were viewed at Leica inverted microscope (Leica, Wetzlar, Germany). Three different sections were chosen for the same animal. There are at least three animals in each group. Three fields were randomly selected for each section, and the mean optical densities (MOD) of positive signals of BMP-2 were analyzed using Image J software. The percentage of MOD of BMP-2 signals were expressed as ratio of MOD of BMP-2 signals vs total signals.
Immunofluorescence For Ki67
The sections were incubated with mouse anti Ki-67 monoclonal anti-body (1:200; Abcam, ab15580, Cambridge, UK), and then incubated with fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG (1:300; Jackson ImmunoResearch Laboratories, West Grove, Pennsylvania, USA). Goat serum was used as blocking solution. Nuclei were stained with Hoechst 33258. Sections were viewed under a laser-scanning confocal microscope (ZEISS LSM 710 META, Oberkochen, Germany). Three different sections were chosen for the same animal. There are at least three animals in each group. The proliferative cells labeled by Ki67 were counted in three different optical fields selected in a random manner, and counted at least 100 cells for each section. The percentage of proliferative cells were expressed as ratio of Ki67-positive cells vs the total number of cells.
Detection of apoptosis assay by TdT-mediated dUTP nick-end Labelling (TUNEL)
TUNEL assays were prepared using an in-situ Cell Death Detection Kit, Fluorescein (Roche, Mannheim, Germany) according to the manufacturer’s instructions. To correlate the TUNEL assay results with the nuclear morphology, the sections were counterstained with hematoxylin. The number of apoptotic cells was counted in 3 randomly selected optical fields (magnification ×400). The samples were by Leica inverted microscope (Leica, Wetzlar, Germany). At least 100 randomly selected cells in each sample were evaluated for apoptosis in the different optical fields (magnification ×400). The results were expressed as the ratio of TUNEL-positive cells to the total number of cells. Each sample was observed at least three times. The TUNEL analysis was performed 3 times for each animal, and the average value was used for the statistical analysis.
Statistical analysis
The results are presented as mean ± standard deviation (SD), and P < 0.05 is considered statistically significant. The data from the blood, HE staining, Sirius red staining, PAS staining, ALP staining immunohistochemical tests and TUNEL were statistically analyzed using one-way analysis of variance (ANOVA). The difference of the bone trabeculae and mineral density between injury side and normal side was analyzed using Student’s t-test.
{kind=link}