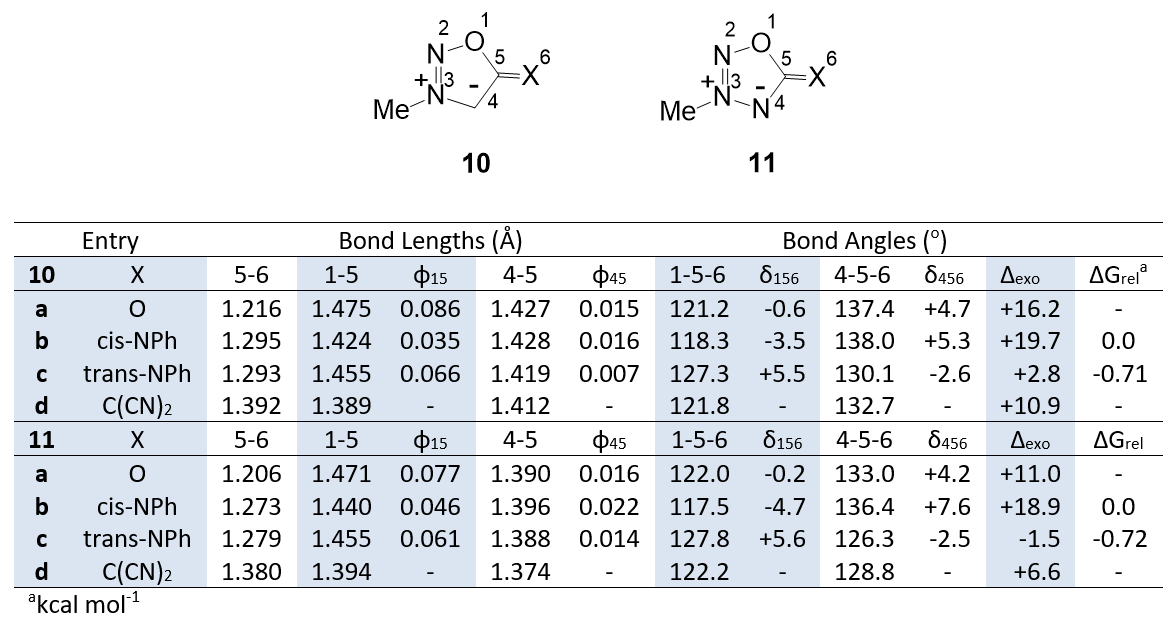
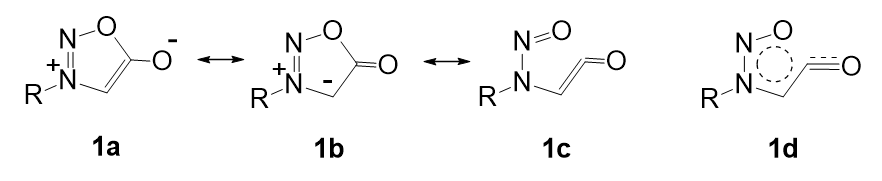
Table 1 shows calculated bond lengths and angles for the mesoionic 1,2,3-oxadiazole and 1,2,3,4-oxatriazole derivatives 10a-d and 11a-d. The configurations of the imines are defined as cis or trans by the relationship of the nitrogen lone pair to the ring C-O or C-NR bond; the use of E and Z nomenclature to define configuration varies with the ring system under consideration and is not a consistent terminology for comparing a series of rings.
Mesoionic rings with an exocyclic C=C(CN)2 group are known [12-15]. We regard the enes 10d and 11d as structures that are not modified by the effects of exocyclic lone pairs and which, therefore, can be used as structural reference points. The effects of lone pairs are measured relative to these geometries. In Tables 1 and 2, φ15 and φ45 are the differences between the bond lengths in structures 10a-c and 11a-c and the corresponding bond lengths in 10d and 11d. Similarly, δ156 and δ456 measure the differences in the corresponding bond angles. The value Δexo is the difference between the exocyclic angles 1-5-6 and 4-5-6 for each structure. ∆Grel are the relative free energies of the imine configurational isomers 10b,c and 11b,c.
The calculated gas phase geometries are in satisfactory agreement with the crystal structures 2, 4 and 8 (Figure 1). Inspection of Table 1 reveals that lone pairs increase the length of the 1-5 bond (φ15) but the effect of a trans lone pair (10c and 11c) is greater and the effect of two lone pairs (10a and 11a) is greatest. A smaller and opposite effect is seen for the 4-5 bond (φ45); here the larger effect is for the cis lone pair and the effects are not additive. The bond angles are also influenced by the configuration of the lone pair. Trans lone pairs increase angle 1-5-6 (δ156) and decrease angle 4-5-6 (δ456); cis lone pairs have the opposite effect. Inspection of the values of ∆exo indicates that trans-imines have the smallest calculated difference between the exocyclic angles and cis-imines have the largest difference. This observation is consistent with the experimentally determined structure 7. It is also significant to note that the trans isomers 10c and 11c are calculated to be the most stable (∆Grel) in accord with structure 8.
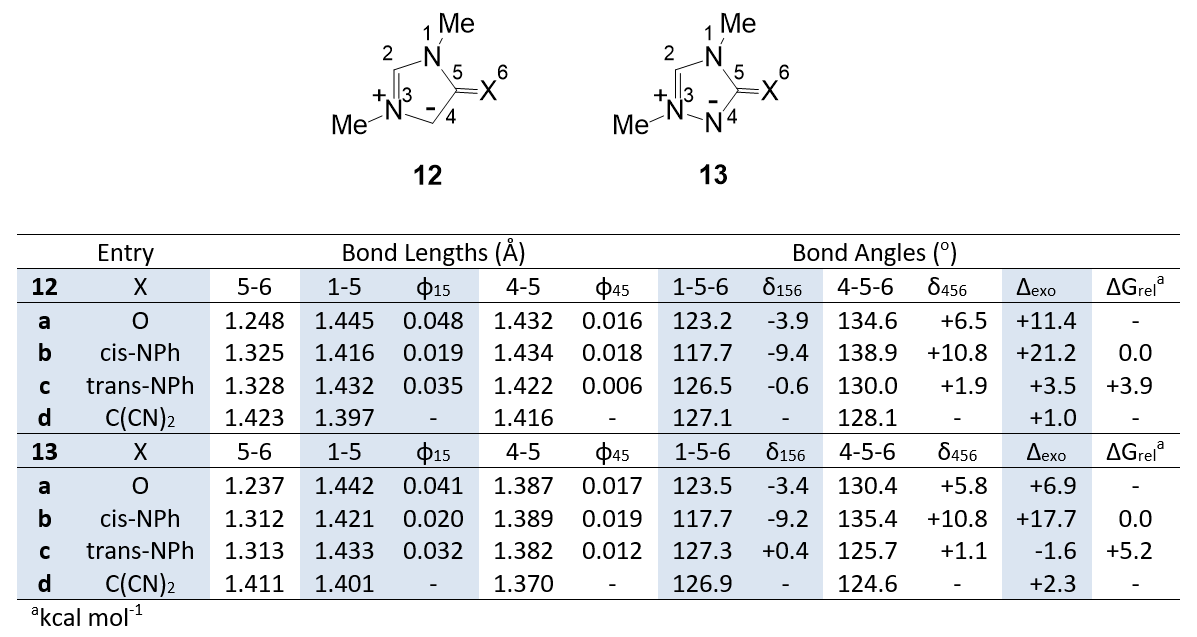
In a parallel study we have calculated the properties of the 1,3-diazoles 12 and the 1,3,4-triazoles 13. Relevant structural properties are shown in Table 2. There is good agreement with the observed geometries of aryl analogues of 12a [16], 12b [10,11] and 13b [8]. Comparison with the data in Table 1 reveals similar trends, with notable differences that can be attributed to differences in C-O and C-NMe bonds. Changes in the ring bonds (φ15 and φ45) show the same trends but the effects are smaller for φ15. Trends in bond angle change with exocyclic group (δ156 and δ456) are comparable in the two series as are the values of ∆exo. However, it is notable that for the imines 12b,c and 13b,c the cis isomers are of lower energy (∆Grel). This is in agreement with the crystal structures of rings 7 and 9 (Figure 2), and can be attributed to steric interactions in the trans isomers. In the trans isomers 12c and 13c the NPh rings are twisted away from planarity (30-40o) indicating energetically unfavourable interactions. Although the cis isomer 13b is fully planar, the Ph group is also twisted in the trans isomer 13c indicating steric interaction with C4H that is absent when replace by N4.
Using the localised bonding model, we rationalise the observed properties in terms of energetically favourable anomeric effects between the exocyclic lone pairs and the antibonding orbitals of the adjacent ring bonds. In the trans configuration 14 the lone pair overlaps with the antibonding orbital of the C-Ẍ bond (n→σCX*) (Figure 3). This stabilising interaction introduces an antibonding element into the C- Ẍ bond resulting in bond lengthening (φ15 positive) while reinforcing the exocyclic C=N bond. The overlap shown in structure 14 is probably increased by increasing the angle 1-5-6 (δ156 positive). In accord with general bond properties, the size of the antibonding lobe on the C5 carbon will be related to the polarity of the bond and will increase as the electronegativity of Ẍ increases. This is consistent with the observation of a greater effect in the C=NPh trans structures 10c and 11c (C-O bond) than in 12c and 13c (C-NMe bond) (Tables 1 and 2).
A similar analysis (Figure 3, Structure 15) accounts for the effect of a cis lone pair on bond lengths and angles. In this case the cis lone pair interacts with the antibonding orbital of the C-C or N-C ring 4-5 bond. Again, the greatest effects are on the more polar N-C bonds (11b and 13b). In the cases where the exocyclic atom is oxygen, both lone pairs influence the geometry. For the 1-5 bonds there is an additive effect but for the 4-5 bonds the combined effect is not significant. For the angles 1-5-6 and 4-5-6 there is a compromise between the effects of cis and trans lone pairs and ∆exo has an intermediate value.
The anomeric effects described here are analogous to other hyperconjugative effects between the antibonding orbitals of polar bonds and adjacent lone pairs, which often account for conformational preferences [17]. Anomeric effects are sometimes attributed to dipolar or steric interactions. In the case of type A mesoionic structures these effects seem unlikely; the derivatives 11, which have similar dipolar and steric characteristics regardless of configuration, have a similar profile to the other derivatives 10, 12 and 13. Within the localized bond model, other orbital interactions obviously influence relative energy and geometry. However, we believe that those shown in Figure 3 dominate. It is also important to recognise that, in addition to lone pairs, other factors influence ring geometries. These include the nature of ring heteroatoms as can be seen from the variation of ∆exo for the dicyano derivatives (10d – 12d, Tables 1 and 2).
We regard the dicyano derivatives as a realistic choice of reference structure. Analogues of this type are known [12-15,18-20], and the exocyclic group C=C(CN)2 is a good lone pair-free electronic analogue of C=O and C=NAr. Unsubstituted derivatives C=CH2 are of some interest for two reasons: (i) mesoionic examples of this type were prepared and fully characterised (with X-ray structures) in 2020 [21]; (ii) the CH bonds may show some hyperconjugative electron donation, in a manner analogous to lone pairs.
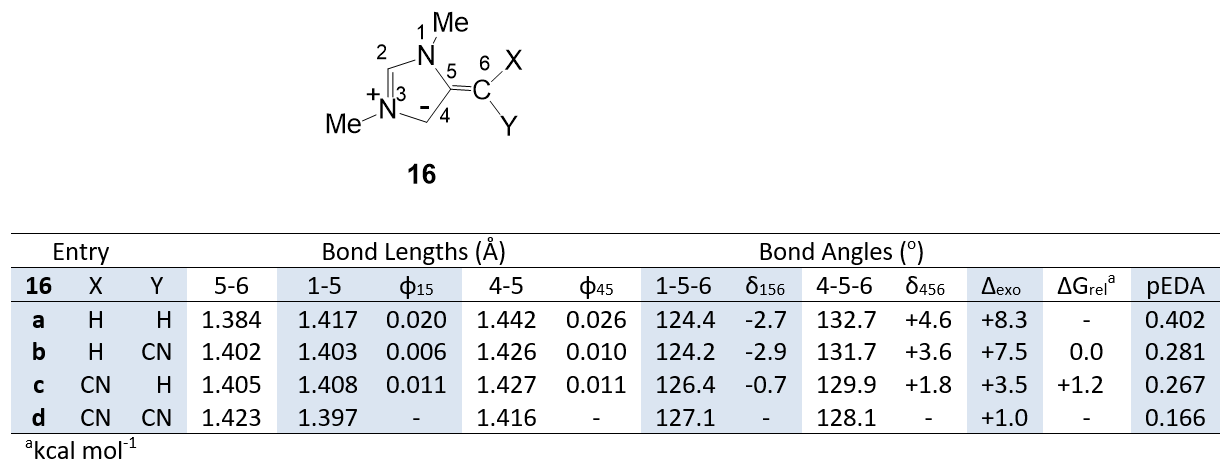
Table 3 compares calculated properties of C=C(CN)2, cis and trans C=CHCN and C=CH2 mesoionic 1,3-diazoles 16. The results reveal some evidence of C-H hyperconjugation, especially by the trans CH bond in structure 16c. Although the effects of the CH bonds in structures 16 are less than those of lone pairs, the trends in bond length and bond angle change are similar, and this suggests an analogous but smaller anomeric effect. The isomer 16c is less stable (∆Grel) than 16b, and this is attributable to a small steric interaction between Me and CN.
The structure of the CH2 derivative 16a is of particular interest since, unlike the other mesoionic rings which are planar, the five-membered ring in structure 16a shows distortion from planarity in both the calculated and X-ray structures (torsion angle 1-5-4-3: calc. 4.4o; obsd 4.2o). This can be attributed to an increased π electron population introducing anti-aromatic character. The index pEDA (pi Electron Donor-Acceptor) is the sum of the populations of the ring pz atomic orbitals minus the aromatic sextet value of six [22]. The pEDA values for the ene structures 16a-d are shown in Table 3. The π-electron ring population of 16a is demonstrably higher than the cyano derivatives 16b-d and also the oxygen and nitrogen derivatives 12a-c (pEDA: 12a 0.230; 12b 0.262; 12c 0.192). Overall the dicyano derivatives have comparable properties to oxygen and nitrogen analogues and are realistic hyperconjugation-free references structures for studying the influence of lone pairs.
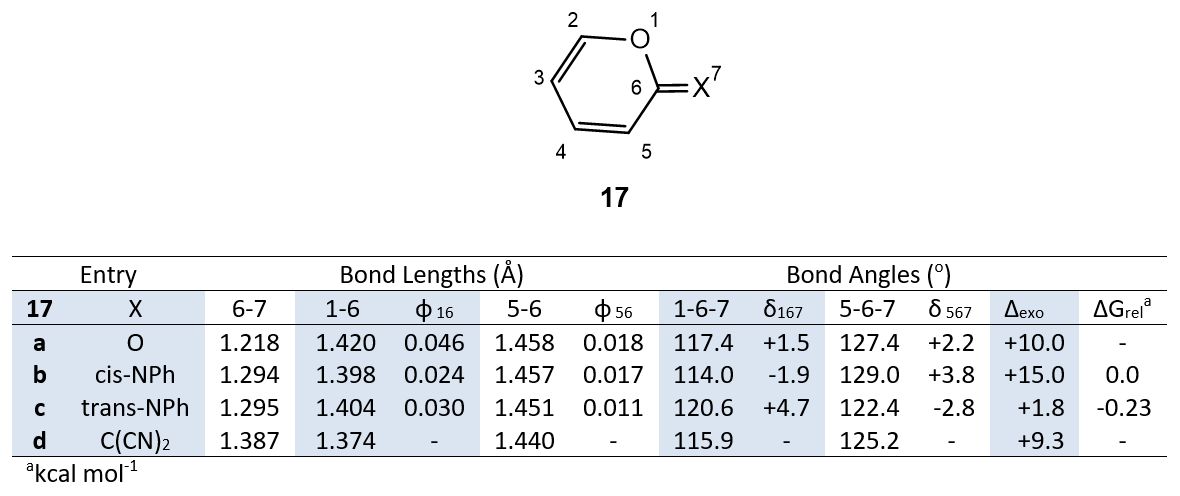
To explore whether similar anomeric effects are found in other heterocyclic rings with exocyclic substituents, we have calculated properties of the pyrone derivatives 17a-d. N,4,6-Triphenypyran-2-imines are known [23,24], and Uncuţa and coworkers, in a detailed NMR study, have shown that in equilibrium mixtures, e.g., 17b,c, the trans (Z) configuration prevails [25].
Examination of Table 4 reveals that the structural changes in the series 17a-d are similar to the trends in Tables 1 and 2. It is noteworthy that the angle difference ∆exo is smallest for the trans isomer 17c (+1.8o) and largest for the cis isomer 17b (+15.0o); this closely parallels the analogous structures 10 and 11 (Table 1). In agreement with experimental data [25], the trans isomer 17c is calculated to be the more stable (∆Grel -0.23 kcal mol-1) (Table 3).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}