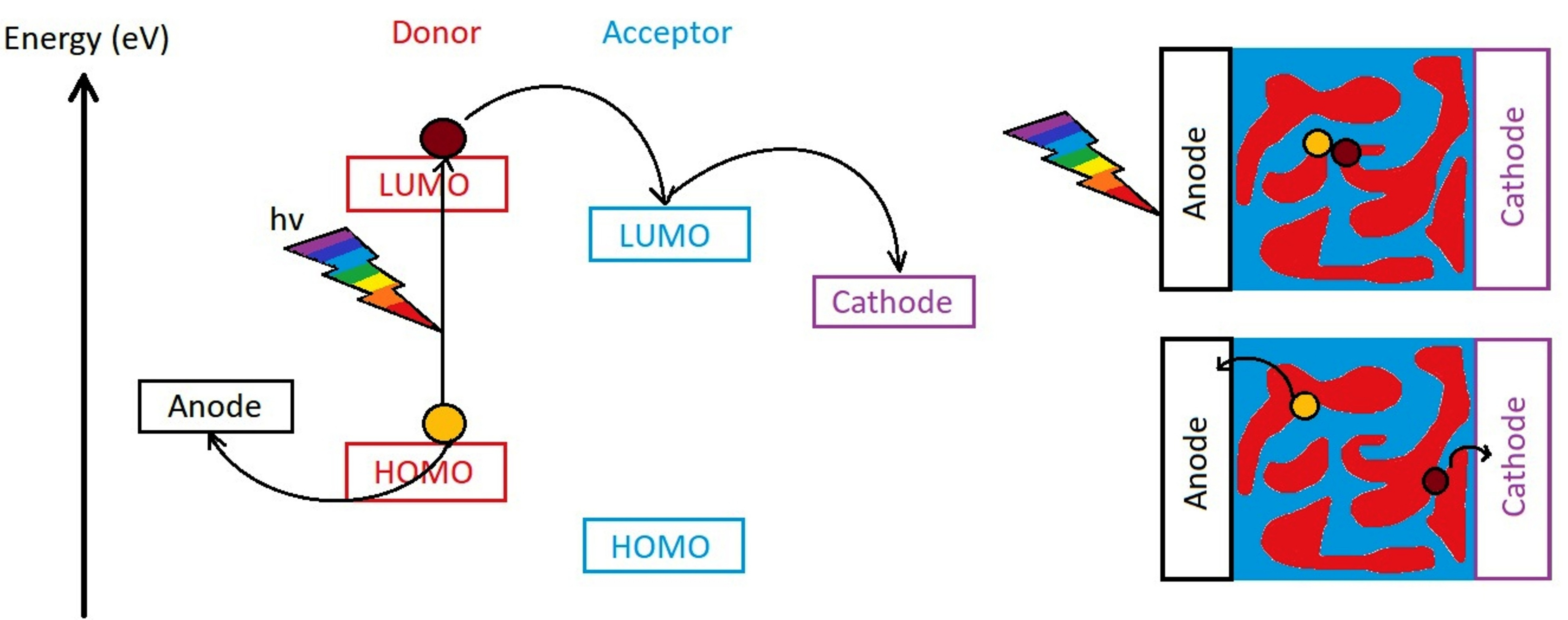
Theoretical analysis on geometries, optoelectronic properties, photovoltaic properties and absorption spectra on six π-conjugated molecules used in organic bulk heterojunction solar cell (BHJ), completed using density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations with Becke’s three-parameter functional and Lee-Yang-Parr functional (B3LYP), hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP) and WB97XD level with 6-311G basis set supported by Gauss View and Gaussian 09 program. LUMO (lowest unoccupied molecular orbital), HOMO (highest occupied molecular orbital), energy gap (Egap) and Voc (open-circuit voltage) and other essential parameters have been investigated to study the effects on substitution of electron-donating and electron-accepting groups to the proposed molecule. These properties determine if the studied compounds can act as good electron donors along with phenyl-C61-butyric acid methyl ester (PCBM) as a suitable electron acceptor.
{kind=link}