Stimuli-responsive behaviour is one of the most fascinating characteristics of smart materials, which endows them with programmability, and thus showing prospective potentials in information storage and encryption, therapeutics as well as biotechnology1–6. Among various external stimuli such as solvent7, mechanical stress8, temperature9 and pressure10, light can be conveniently and rapidly controlled to actuate molecules with superior spatiotemporal accuracy11–14. Thus far, lots of photo-responsive materials together with respective mechanisms have been proposed, including molecular photoswitches (e.g. azobenzene, spiropyran and diarylethene)15–18, photoactivated phosphorescence via 3O2 consumption19–22 and photoinduced radical materials23–26. Nevertheless, most of them, especially those undergo photochemical reactions, experience colour change of appearance during photoluminescence (PL) transformation, which limits their applications in information encryption. Furthermore, they normally exhibit confined PL colour changing ranges. Meanwhile, their syntheses are generally troublesome. Therefore, the development of intelligent and easily accessible photo-responsive materials with a wide range of colours still poses a significant challenge.
It has been widely acknowledged that molecular packing plays an essential role in organic solid-state emission27, for which slight changes in molecular arrangement can lead to gigantic variations in photophysical properties28. For instance, different polymorphs of a compound often exhibit completely different emission behaviors29,30. Therefore, controlling the aggregation state offers a promising approach to achieve diverse luminescence. However, the challenge lies in the fact that current methods for obtaining distinct aggregation states are primarily limited to artificials means, such as single crystal cultivation using various solvents. The spontaneous transformation between target aggregation states is rarely achieved31–33. Consequently, there is a pressing need to develop materials that can change their aggregation state in response to external stimuli, particularly light. This advancement would greatly contribute to the development of intelligent luminescent materials.
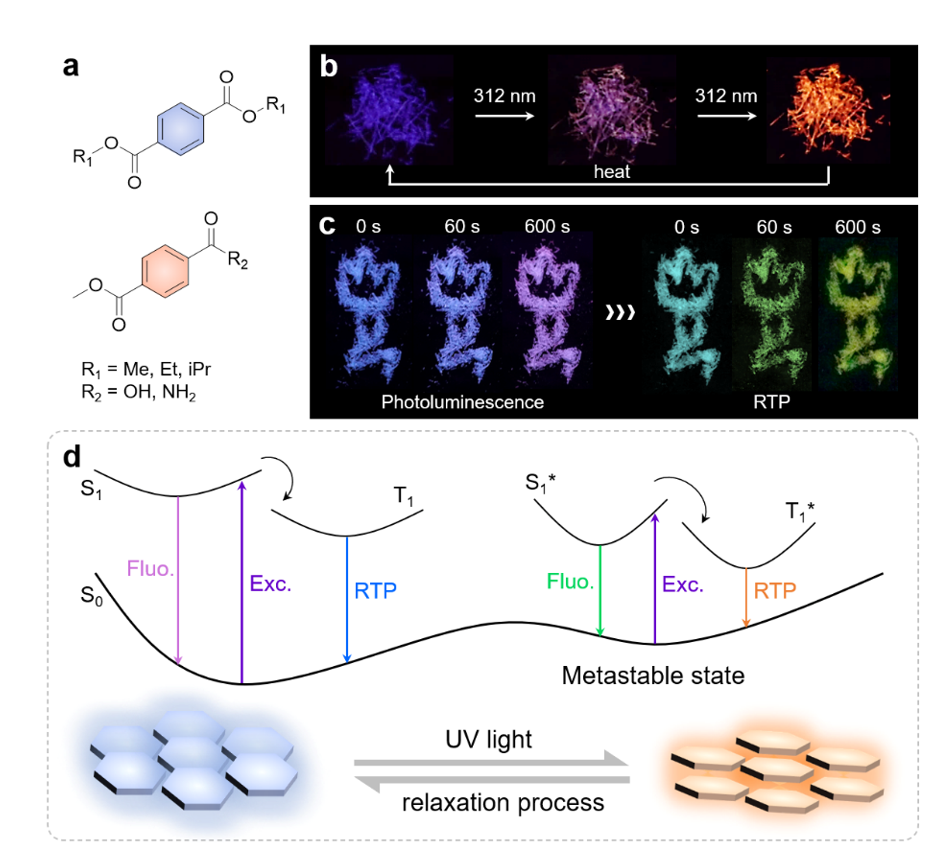
Here we report a surprising discovery of photoinduced PL transformation from crystalline dimethyl terephthalate (DMTPA) (Scheme 1a), which has been previously reported as a compound with crystallization-induced phosphorescence and clustering-triggered emission (CTE) features34,35. It is newly found that DMTPA crystals undergo an intriguing photoinduced PL colour variation from dark blue to purple and then to bright orange upon exposure to UV light. Furthermore, after heat treatment or solvent fuming, the crystal can restore to its original blue emission (Scheme 1b). Remarkably, while the PL colour switching process goes immediately, the orange emission state can maintain for over 3 months under ambient conditions, demonstrating outstanding stability. Further investigation reveals that subtle molecular rearrangement in crystals results in a metastable aggregation state with a lower energy gap from the thermodynamically stable state. This photo-luminochromism (PLC) phenomenon is driven by UV irradiation (Scheme 1d) and can be reversed upon sufficient relaxation. Building upon these findings, we designed and synthesized additional analogues to verify the abovementioned mechanism (Scheme 1a), leading to the development of several photo-responsive PL materials with diverse photophysical properties. For instance, by replacing one ester group with carboxyl capable of forming strong hydrogen bonds, monomethyl terephthalate (MMTPA) crystal exhibits photoinduced colour changing in persistent room temperature phosphorescence (p-RTP) (Scheme 1c). The discovery of such reverse photo-luminochromic systems through molecular rearrangement will contribute to the facile construction of smart luminescent materials for practical advanced technical applications.
Photo-luminochromic behaviors of DMTPA crystals
DMTPA crystals demonstrate astonishing PL colour transformation under 312 nm UV irradiation, which turns from dark blue to purple in 10 s and continuously to bright orange in ~ 5 min (Fig. 1a and Supplementary Video 1), while the appearance of the crystals remains unchanged. Notably, UV irradiation with other wavelengths (e.g. 330 nm) can also trigger this process. In situ emission spectra in Fig. 1b illustrate the dynamic change in PL of DMTPA crystals. Prior to UV exposure, the spectrum of DMTPA crystals shows a single peak at ~ 420 nm, corresponding to blue emission with CIE (Commission Internationale de I’Eclairage) coordinate of (0.17, 0.09) (Fig. 1c). After 3 s of irradiation, three new peaks appear at 595, 650 and 725 nm and gradually increase in intensity. Additionally, two inconspicuous peaks at 515 and 550 nm show slight increments, while the original peak at ~ 420 nm significantly descends. Correspondingly, the resulting emission colour shifts towards the purple region with CIE coordinate at (0.22, 0.13). This transformation process continues as the irradiation progresses and reaches its final state at ~ 1000 s, with vibrant orange-red emission at CIE coordinate of (0.50, 0.39). At this point, the peak at ~ 420 nm becomes negligible while the newly generated peaks in the redder region are predominant. Importantly, this emitting property can persist for more than 3 months under ambient conditions, implying outstanding stability of the newly generated emissive centres. Moreover, the invariant 1H and 13C NMR, EPR and HPLC spectra of DMTPA before and after UV irradiation (Supplementary Figs. 1, 2, 15 and 16) strongly indicate the absence of photochemical reactions. The consumption of 3O2 is also ruled out since the same PLC phenomenon is observed in vacuum (Supplementary Fig. 17). It is thus naturally deduced that some physical changes are responsible for the PL transformation.
Significantly, the delayed PL spectra of DMTPA crystals are basically consistent with the prompt ones (Supplementary Fig. 18). This result as well as the time-resolved measurements discloses the millisecond (ms)-scale lifetimes for nearly all peaks ranging from 420 to 725 nm (Fig. 1d and Supplementary Fig. 19a). Meanwhile, nanosecond (ns)-scale lifetime is only detected at ~ 420 and 515 nm, while none is present at the photo-generated ones (595, 650 and 725 nm). Above results are indicative of both fluorescence and phosphorescence dual emission at ~ 420 and 515 nm, and pure phosphorescence for other peaks (Supplementary Figs. 19b,c). Furthermore, prompt and delayed emission spectra of 10–5 M DMTPA/2-MTHF solution at 77 K reveal the fluorescence and phosphorescence for monomeric DMTPA at 302 and 420 nm, respectively (Supplementary Fig. 20). It is also noted that DMTPA dimers fluoresce UV at around 374 nm35. Therefore, it can be inferred that the broad peak around ~ 420 nm comprises monomeric phosphorescence and aggregate fluorescence of DMTPA, while the photo-generated ones at longer wavelengths are attributable to phosphorescence from clustered aggregates40. Consequently, the photoinduced PL colour variation, namely PLC, is highly correlated with the emerging DMTPA clusters in response to external UV stimulus. This conclusion is further supported by the absorption of DMTPA crystals (Supplementary Fig. 21), in which a peak at 350 nm arises as the exposure time increases, implying newly generated aggregates with extended conjugation.
In addition to the emerging peaks upon UV exposure, there is a noticeable decline in PL intensity at ~ 420 nm (Fig. 1b). To gain more information, excitation spectra of DMTPA crystals were measured (Fig. 1e). While an intense peak at exactly 420 nm is observed with the emission wavelengths of 595 and 650 nm, the optimal excitation for the 725 nm emission is 650 nm. This result suggests a potential tandem energy transfer process (Fig. 1f). During the PLC process, emission at 420 nm can be readily absorbed by clusters emitting at 595 and 650 nm, and subsequently, emission at 650 nm is further utilized by the clusters luminescing at 725 nm. Such energy transfer hypothesis gains support from the emission spectrum with excitation wavelength of 420 nm, which can be deconvoluted into four peaks locating at 591, 632, 659 and 706 nm (Supplementary Fig. 22), basically consistent with those excited by 312 nm UV light. Moreover, as aggregates accumulate in response to prolonged UV stimulus, the consumption of emission at 420 nm will progressively increase, thus leading to the attenuation at ~ 420 nm.
Understanding the mechanism underlying photo-luminochromism
As discussed above, the generation of new aggregates responding to UV stimulus is responsible for the PLC process. These new aggregates, however, do not originate from photoinduced clustering of discrete molecules, but from the transition of original aggregates. To provide more compelling evidences, we conducted characterizations focusing on variations in intermolecular interactions of DMTPA crystals.
Firstly, DMTPA crystals before and after photo-irradiation were characterized by X-ray diffraction (XRD) technology (Fig. 2a). The exposure time under 312 nm UV light was extended to 12 h to amplify the variations detectable. While the diffraction peaks corresponding to (111), (112) and (002) faces disappeared, a new weak peak corresponding to (119) diffraction emerged, suggesting the adjustments in molecular alignment within crystals. Meanwhile, Raman spectroscopy is known for its sensitivity in detecting slight morphological variations of molecular conformations owing to its ultra-surface-sensitivity36,37. As demonstrated in Fig. 2b, the relative Raman intensity of the peak at 3085 cm–1, which corresponds to stretching vibration of aromatic C–H bond, remains intact after 5 min of 312 nm UV irradiation, despite the PL transformation has simultaneously occurred. However, after continuous irradiation for 12 h, the relative intensity of it remarkably descends, indicating changes in intermolecular interactions, particularly π-π interactions. These results seem indicate that the molecular rearrangement is too minuscule to be perceived early in the PLC process, while is accumulated and become detectable with prolonged irradiation. Additionally, the peak at 87 cm–1 is shifted to 96 cm–1 upon continuous UV stimulus (Fig. 2b), implying increased restrictions on molecular motions38. Such restrictions effectively suppress nonradiative decay processes and stabilize triplet excitons, providing a rational explanation for the phosphorescence properties observed in DMTPA crystals after undergoing photoinduced changes. Further in situ FTIR spectra focusing on the skeletal vibrations of aromatic C = C bonds disclose an increasing trend in characteristic peaks at 1608, 1535 and 1503 cm–1 with prolonged exposure time (Fig. 2c)38, which is consistent with the result obtained from Raman analysis, suggesting enhanced π-π interactions upon UV irradiation.
To probe the structure change of DMTPA crystals, single crystal XRD analysis was further performed (Fig. 2d and Supplementary Figs. 23a,b). While there is no face-to-face π-π stacking among molecules due to the excessive distance (Fig. 2d, top), abundant edge-to-face π-π interactions are found, thanks to the tight molecular packing of DMTPA (Fig. 2d, bottom)39,40. Such interactions are duly verified by the theoretical calculation of noncovalent interactions (NCI) analysis (Fig. 2d and Supplementary Fig. 23c). Although not as strong as the face-to-face interactions, these widespread edge-to-face π-π interactions, together with other short contacts, form an effective through-space conjugation (TSC) network that promotes much redder emissions. These intermolecular interactions and TSC prevents a drastic transformation in the crystal structure upon irradiation, allowing subtle changes in molecular alignment during the PL transformation, as confirmed by the in situ single crystal analysis. The dihedral angle between two DMTPA molecules involved in edge-to-face π-π interactions gradually decreases during the PLC process (Supplementary Fig. 24), and the bond lengths of C = O∙∙∙H–C short contacts between longitudinally arranged molecules shorten from 2.773 to 2.770 Å in 5 min, and then to 2.762 Å in 12 h (Supplementary Fig. 25). These observations indicate an enhancement of π-π interactions, which is consistent with the FTIR results and further validated by the shortened distances of the edge-to-face π-π interactions (Supplementary Fig. 26).
Notably, not all intermolecular interactions are strengthened in response to photo-stimulus. The C = O∙∙∙C and C = O∙∙∙H–C short contacts between transversely arranged DMTPA molecules gradually lengthen from 3.183 and 2.815 Å to 3.185 and 2.820 Å, respectively (Supplementary Fig. 25). The crystal unit cell contracts during the transformation, as evidenced by the reduction in the edge lengths while the density increases (Supplementary Table 1). These changes along with enhanced TSC account for the long-wavelength emission of the rearranged aggregates. Despite being subtle, these photoinduced variations are sufficient to cause significant PL transformations of the aggregates due to amplification effects from the dense and orderly periodic TSC network.
To further investigate varied intermolecular interactions, growth morphology modeling utilizing the Bravais-Friedel Donnay-Harker (BFDH) method was performed based on 2θ scans and single crystal XRD data using the Mercury package41. The drastic decrease in intensity observed at 2θ of 25.1° indicates a variation in molecular arrangement on the (002) face (Fig. 2a), which corresponds to C = O∙∙∙C and C = O∙∙∙H–C short contacts that have been shown to change (Fig. 2e and Supplementary Fig. 25). The weakening of these contacts is also evident in the NCI analysis plot (Supplementary Fig. 27). Similarly, the depressed peak at 2θ of 19.7° also implies the change of molecular arrangement along the (111) face, corresponding to the edge-to-face π-π interactions and C = O∙∙∙H–C contacts between longitudinally arrayed molecules (Supplementary Fig. 28). These results elucidate variations in multiple interactions around DMTPA molecules in response to photo-stimuli, providing additional evidence for photoresponsive molecular rearrangement.
Furthermore, TD-DFT theoretical calculations were performed on DMTPA trimers extracted from the single crystal structure. Before photo-stimulus, two intersystem crossing (ISC) channels from S1 to T7 are observed, with an energy gap of 0.308 eV (Supplementary Fig. 29). HOMOs show through-space edge-to-face π-π interactions distributed across the trimer, whereas the LUMO is mainly located at two molecules, indicative of a partial charge transfer feature (Fig. 2f). After 312 nm UV irradiation, the trimer exhibits four ISC channels, still from S1 to T7, with a decreased energy gap of 0.306 eV (Fig. 2g and Supplementary Fig. 30). Increased ISC channels and narrowed energy gaps (ΔEST) are favorable for the phosphorescence, which is consistent with the experimental results.
Derived from above discussions, the mechanism underlying PLC of DMTPA crystals can be concluded as follows: External photo-stimulus drives the subtle molecular rearrangement, which is amplified by the compact and orderly periodic TSC network throughout the DMTPA crystal lattice, thereby forming new clustered emitters with distinct emissions. Additionally, moderate short contacts between transversely arranged molecules from ester groups are also necessary for empowering molecular reorganization under mild stimuli. Moreover, the crystalline nature of DMTPA is necessary for PLC. While the liquid nitrogen quenched DMTPA solid (also crystalline) show evidently decreased PLC property, its ground powder (almost amorphous) demonstrates neglectable change upon UV irradiation (Supplementary Fig. 31).
Universality of photo-luminochromism and mechanism verification
As elaborated in the proposed mechanism, through-space electron interactions within the crystal play an essential role in the formation of new emissive clusters and amplification of the effects of subtle molecular rearrangement. To verify it, we investigated diethyl terephthalate (DETPA) and diisopropyl terephthalate (DiPTPA) with larger alkyls, which are effective in tuning the π-π interactions. Despite both compounds demonstrate variations in PL spectra upon UV irradiation free of any photochemical reactions (Supplementary Figs. 3, 4, 32 and 33), only DETPA crystals show colour-changing PL, transitioning from blue to purple after 10 min of irradiation, whereas the emission of DiPTPA crystals gradually fades from bright light blue (Fig. 3a). Concretely, other than the decrease in intensity at ~ 440 nm, three peaks at 605, 665 and 730 nm emerge in the prompt PL for DETPA crystals upon irradiation (Fig. 3b), whereas only the attenuation at ~ 420 nm is observed for DiPTPA crystals (Fig. 3c). Moreover, ms-scale lifetime measurement reveals the predominant triplet feature for all peaks (Supplementary Figs. 34–37), while the emerging ones exhibit no ns-scale components (Supplementary Fig. 38), illustrating their RTP nature. Furthermore, variations in XRD patterns confirm the occurrence of molecular rearrangement in both crystals irradiation (Supplementary Fig. 39), which is further verified by the in situ single-crystal analysis (Supplementary Figs. 40 and 41 and Supplementary Tables 2 and 3).
However, although both DETPA and DiPTPA crystals are proved to undergo molecular rearrangement by XRD analysis, the through-space electron interactions in the latter are not enough to provide an amplification effect due to the strong steric hindrance. As demonstrated in Fig. 3f, widespread face-to-face π-π interactions are observed in DETPA crystals, whereas there is no effective π-π stacking among DiPTPA molecules. Furthermore, Figs. 3d,e show the FTIR spectra of these crystals in the skeletal vibration region of aromatic ring. The characteristic peaks of DETPA crystals rise evidently after 12 h of UV stimulus, illustrating the photoinduced enhancement of π-π interactions (Fig. 3d). In contrast, those of DiPTPA crystals depicts tiny changes with varying exposure time (Fig. 3e), suggesting inappreciable variation of π-π interactions. The lack of efficient π-π stacking arising from the large steric hindrance of isopropyls hinders the molecular aggregation, thereby suppressing the generation of new clusters and preventing changes in PL colour. In this sense, the amplification effect deriving from the aggregation and through-space electron interactions play an indispensable role in generating new clustered emissive species, without which subtle molecular rearrangements are insufficient to prompt PLC process.
Nevertheless, excessive intermolecular interactions can hinder molecular rearrangement. For example, dimethyl 2,6-naphthalenedicarboxylate, despite owning a similar crystal structure to DMTPA (Supplementary Fig. 42), only demonstrates unremarkable PL colour transformation from dark purple to violet even after 20 min of UV exposure, accompanying negligible newly generated PL peaks (Supplementary Fig. 43). Subtle molecular rearrangement can be noticed after 12 h of accumulation (Supplementary Figs. 44 and 45 and Supplementary Table 4), the process, however, is significantly more difficult compared to that observed in DMTPA crystals.
It should be stressed that the amplification effect empowered by through-space electron interactions is not the only prerequisite of PLC. The strength of short contacts between transversely arranged molecules is identified as another factor influencing the molecular rearrangement. To explore this further, MMTPA and terephthalic acid (TPA) crystals are chosen for comparison, for which strong hydrogen bonds are expected. Impressively, upon 10 min of 312 nm UV irradiation, MMTPA crystals not only exhibit PL colour changing from dark blue to bright magenta, but also display tunable afterglow varying from cyan to green and then to yellow (Figs. 4a,b). The emission of MMTPA crystals exhibits ultralong lifetimes (Supplementary Fig. 46), indicating their p-RTP nature, which are attributed to the hydrogen bonding stabilized triplet excitons. To the best of our knowledge, this is the first example showing p-RTP afterglow with remarkable PLC through the molecular rearrangement mechanism. Correspondingly, the peaks at ~ 430 and ~ 530 nm in the prompt PL spectra descend as exposure time increases, while those at 600, 655 and 720 nm simultaneously increase (Fig. 4b), accompanying CIE coordinate shifting from (0.20, 0.19) to (0.37, 0.24) (Fig. 4c).
Distinct from DMTPA, the peak at ~ 530 nm in the delayed PL spectra remain predominate after UV irradiation (Fig. 4d), thus leading to p-RTP located in the upper right region of the CIE diagram (Fig. 4c). Meanwhile, both prompt and delayed emissions at 600, 655 and 720 nm also contribute relatively small fractions to the whole PL compared to those of DMTPA and DETPA, suggesting that stronger short contacts somehow inhibit the PL transformation process. The photoinduced molecular rearrangement mechanism is still applicable to the PL transformation of MMTPA crystals and can be verified through similar characterizations, including in situ FTIR spectra (Fig. 4e), XRD patterns (Supplementary Fig. 47) as well as the single-crystal analysis (Supplementary Fig. 48 and Supplementary Table 5). Therefore, by introducing stronger hydrogen bonds, MMTPA crystals can also demonstrate PLC phenomenon and moreover with distinct p-RTP colour transformations, thus shedding lights on the fabrication of novel multicolor p-RTP smart materials with PLC property. In contrast, TPA powder crystals show hardly any change in PL after 10 min of 312 nm UV irradiation (Fig. 4a, Supplementary Fig. 49), along with virtually unaltered XRD patterns and FTIR spectra (Supplementary Fig. 50). However, TPA crystals exhibit even longer p-RTP than that of MMTPA crystals (Supplementary Fig. 51).
Clearly, as the ester groups are gradually replaced by carboxyl units, the crystals tend to generate much longer and more efficient p-RTP with attenuated PLC behaviors. To uncover the reason behind this evolution, their single-crystal structures were compared (Fig. 4f). In DMTPA crystals, molecules are loosely packed due to the weak short contacts between ester groups, making them more prone to rearrange in response of external photo-stimulus, thus readily to proceed PLC. However, such weak intermolecular interactions cannot fully restrain nonradiative transitions, thus leading to relatively short RTP. For MMTPA, two molecules form an integral dimer through multiple hydrogen bonds between adjacent carboxyl groups (Supplementary Fig. 52). Thanks to the ester groups at each end of the dimers, they can arrange in a manner similar to DMTPA molecules, thus retaining the ability of PLC. Meanwhile, the introduction of hydrogen bonds is favorable to conformational rigidity, which effectively stabilize triplet excitons and further lead to photo-luminochromic p-RTP emission. In contrast, TPA molecules are tightly connected end to end through multiple hydrogen bonds, π-π interactions and other short contacts, forming a compactly packed planar structure with ample interlayer O = C∙∙∙C interactions (Supplementary Fig. 53)42, which effectively prohibit molecular rearrangement upon photo-irradiation while promoting p-RTP emission. These results strongly imply that moderate short contacts between transversely arranged molecules is a key factor empowering molecular reorganization in response to photo-stimulus. This notion is further supported by the presence of PLC in methyl 4-(aminocarbonyl)benzoate crystals and absence of PLC in terephthalamide crystals (Supplementary Fig. 54).
As indicated above, the amplification effect facilitated by through-space electron interactions plays a crucial role in PLC process, which enables even subtle molecular rearrangements to trigger the formation of newly clustered emissive species. Additionally, moderate short contacts are necessary for allowing such molecular reorganization in response to mild photo-stimulus. Inspired by this mechanism, it is feasible to regulate the colour range and response time of PLC by appropriately adjusting the moieties. Moreover, the integration of p-RTP with PLC becomes achievable by introducing stronger short contacts. These approaches ensure the rational design of more functional photo-responsive PL materials.
Reversibility and applications
As exhibited in Fig. 5a, photo-responsive DMTPA crystals can resume blue emission after heating for 10 min at 80 oC in air, and the PL transformation process towards orange emission will be repeated with further 312 nm UV irradiation, illustrating excellent reversibility. On the other hand, since the PLC property of these compounds are highly dependent on the crystal structure (Supplementary Fig. 31), grinding is another way to reawaken the blue emission from DMTPA crystals. Upon solvent fuming and consequent UV irradiation, the ground powders show orange PL again. Furthermore, the emission spectra during the PL colour variation-recovery processes disclose the almost identical profiles of the photo-irradiated samples, while the main peaks of ground or heated crystals hypsochromically shifted with comparison to original DMTPA crystals (Fig. 5b). This blue-shift should be ascribed to the destruction of crystal structure that attenuates conformational rigidity, causing the decrement of monomeric phosphorescence at ~ 420 nm. Additionally, the orange PL of transformed DMTPA crystals can spontaneously revert to purple when placed at ambient conditions for over 3 months. This reversal process indicates that the molecular rearrangement from orange-emitting clusters to blue-emitting ones is thermodynamically permissible and that the former is a metastable state generated in response to external UV stimulus (Fig. 1d).
The unique PLC behaviors of DMTPA crystals endow them promising applications in smart encryption ink and information storage. For instance, a 5×4 cm2 butterfly pattern was printed on a filter paper through simple screen printing method with a mixture of commercially available aloe gel and DMTPA/EA (right wing) or DiPTPA/EA (left wing) solutions (Fig. 5c). The initial PL from both wings is similar. However, upon exposure to 312 nm UV light for 1 min, the PL from the right wing becomes purple, and further turns to conspicuous orange-red with increasing exposure time of 3 min (Fig. 5d). In contrast, the PL of the left wing gradually fades away. This experiment clearly demonstrates the feasibility of using DMTPA as intelligent encryption ink.
However, the crystalline nature and thus non-deformability and fragility of DMTPA crystals limited their applications. To overcome this limitation, we combined DMTPA crystals with polyacrylamide (PAM) hydrogel to create a flexible and reusable material featuring photo-responsive PL transformation for information storage and erasure. As schemed in Fig. 5e, tiny DMTPA crystals were dispersed into an aqueous PAM solution to prepare the hydrogel that emits blue PL under 312 nm UV light. By exposing specific areas through a mask for ~ 5 min followed by removal of the mask, red emission is observed in the exposed part while the rest of the hydrogel remains blue, thus enabling information storage. The input pattern can be easily erased by further UV irradiation to transform the entire hydrogel into a red-emitting state. After erasing the information, the hydrogel can be reused by heating it at 80 oC for 10 min to restore its blue PL. The practical application of this hydrogel is shown in Fig. 5f, demonstrating its potential prospects in intelligent information carriers and flexible devices utilizing the intriguing PLC phenomenon.
{kind=link}