Backgound
Mixed models are used to correct for confounding due to population stratification and hidden relatedness in genome-wide association studies. This class of models includes linear mixed models and generalized linear mixed models. Existing mixed model approaches to correct for population substructure have been previously investigated with both continuous and case/control response variables. However, they have not been investigated in the context of extreme phenotype sampling (EPS), where genetic covariates are only collected on samples having extreme response variable values.
Methods
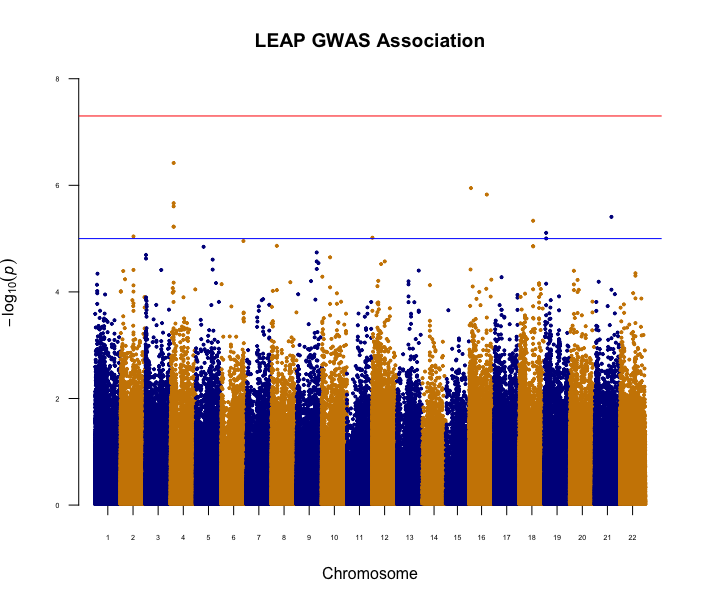
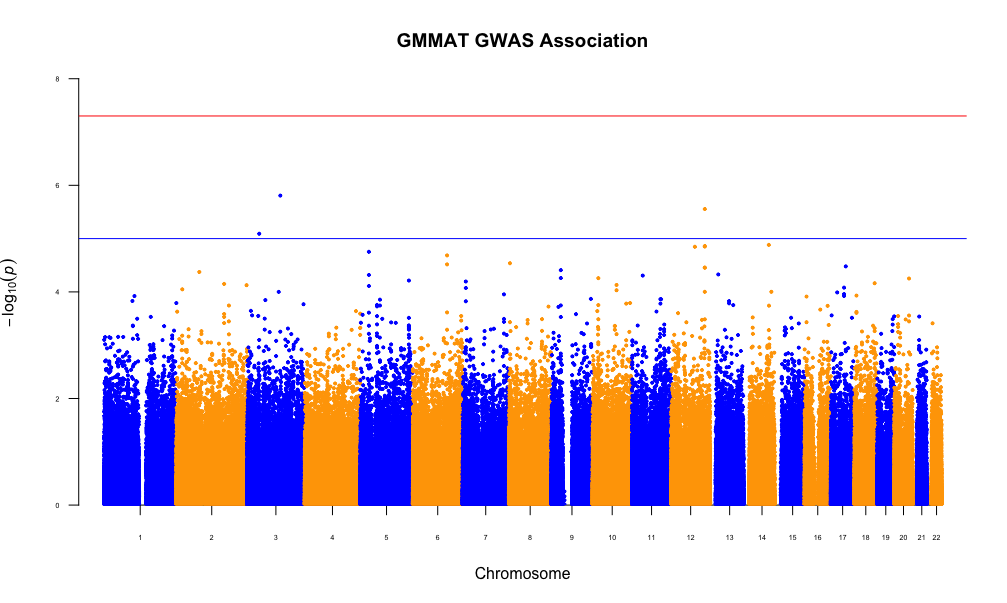
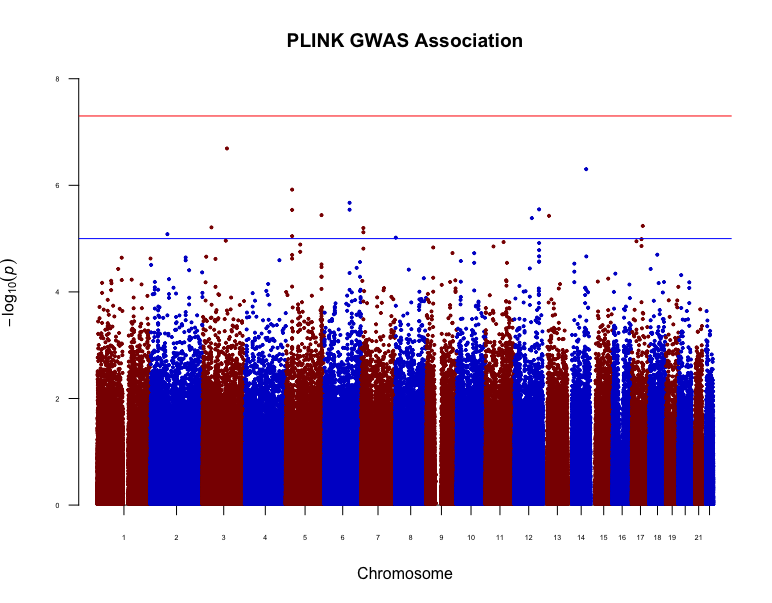
In this work, we compare the performance of existing binary trait mixed model approaches (GMMAT, LEAP and CARAT) on EPS data. Since linear mixed models are commonly used even with binary traits, we also evaluate the performance of a popular linear mixed model implementation (GEMMA). We use simulation to estimate the type 1 error of all approaches under confounding due to population stratification. We also apply all methods to a real dataset from a Québec, Canada, case-control study that is known to have population substructure.
Results
Our simulation results show that for a common candidate variant, both LEAP and GMMAT control the type 1 error rate. We observe similar type 1 error control with the analysis on the Québec dataset. However, for rare variants the false positive rate remains inflated even after correction with mixed model approaches.
Conclusions
The methods compared in this study do not perform equally well. Therefore, when data are from an EPS study, care should be taken to ensure that the models underlying the methodology are suitable to the sampling strategy and to the minor allele frequency of the candidate SNPs.

{kind=link}
{kind=link}
{kind=link}