2.1. Materials
Phosphate-buffered saline (PBS), paraformaldehyde, potassium chloride, sodium bicarbonate, sodium chloride, monopotassium phosphate, magnesium dichloride hexahydrate, ammonium carbonate, calcium chloride dihydrate, hydrochloric acid, bovine bile, porcine pepsin, porcine pancreatin and p-toluene-sulfonyl-L-arginine methyl ester (TAME) were obtained from Merck (Amsterdam, Netherlands). Laemmli sample buffer (2×) was obtained from Bio-Rad (Lunteren, The Netherlands). Trifluoroacetic acid, dithiothreitol and iodoacetamide were obtained from Sigma Aldrich (Zwijndrecht, The Netherlands), and sequencing grade trypsin was obtained from Boehringer Mannheim (Mannheim, Germany). Precast SurePAGE MOPS-Tris PAGE gels were obtained from Genscript (Rijswijk, The Netherlands). Alexa-fluor 594-conjugated wheat germ agglutinin was obtained from Thermo Fisher (Bremen, Germany).
2.2. Particles
Green fluorescent (Ex 441, Em 485) and nonfluorescent polystyrene (PS) MNPs (50 nm, 100 nm, 200 nm, 500 nm, 1000 nm) were obtained from Polysciences (Hirschberg an der Bergstraße, Germany). Green fluorescent and nonfluorescent carboxyl- and amine-modified PS MNPs (100 nm) were obtained from Magsphere (Pasadena, USA). Throughout the rest of the manuscript, the particles will be referred to as shown in Table 1.
Table 1: MNPs used in this study with the respective abbreviations
|
Particle
|
Abbreviation
|
|
Polystyrene, 50 nm
|
PS50
|
|
Polystyrene, 100 nm
|
PS100
|
|
Amine-modified polystyrene, positive charge, 100 nm
|
PS100 (+)
|
|
Carboxyl-modified polystyrene, negative charge, 100 nm
|
PS100 (-)
|
|
Polystyrene, 200 nm
|
PS200
|
|
Polystyrene, 500 nm
|
PS500
|
|
Polystyrene, 1000 nm
|
PS1000
|
2.3. In vitro gastrointestinal digestion of MNPs
For the in vitro gastrointestinal digestion, the INFOGEST-2 protocol (31) was used with minor modifications. Briefly, the digestion protocol consists of 3 steps, namely, the sequential addition of simulated salivary fluid (SSF), simulated gastric fluid (SGF) and simulated intestinal fluid (SIF) to the sample of interest, representing the oral, gastric and intestinal digestive phases, respectively (table 2).
Table 2: Composition of digestive fluids
|
Component
|
Simulated salivary fluid (SSF) (pH = 7.0)
|
Simulated gastric fluid (SGF) (pH = 3.0)
|
Simulated intestinal fluid (SIF) (pH = 7.0)
|
|
KCl
|
30.2 mM
|
6.9 mM
|
6.8 mM
|
|
NaHCO3
|
27.2 mM
|
25.0 mM
|
85.0 mM
|
|
NaCl
|
-
|
47.2 mM
|
38.4 mM
|
|
KH2PO4
|
7.4 mM
|
0.9 mM
|
0.8 mM
|
|
MgCl2. (H2O)6
|
0.3 mM
|
0.1 mM
|
0.3 mM
|
|
(NH4)2CO3
|
0.1 mM
|
0.5 mM
|
-
|
|
CaCl2(H2O)2
|
1.5 mM
|
0.15 mM
|
0.6 mM
|
|
Pepsin
|
-
|
0.8 mg/ml
|
-
|
|
Bovine Bile
|
-
|
-
|
4.0 mg/ml
|
|
Porcine Pancreatin
|
-
|
-
|
14.0 mg/ml
|
After dissolution of all the salts, the pH of the SSF, SGF and SIF was adjusted to 7.0 ± 0.5, 3.0 ± 0.5, and 7.0 ± 0.5 for each fluid, respectively, using 1 mM HCl, and subsequently, all fluids were autoclaved. Prior to in vitro gastrointestinal digestion, 1000 U/ml pepsin was added to SGF, while 4 mg/ml bile and 100 U/ml pancreatin were added to SIF. Bile and pancreatin were dissolved by incubation at room temperature (RT) while rotating head-over-heels for 1 h (at 25 RPM), and pepsin was dissolved by head-over-heels rotation for 5 min at RT. After addition of the proteins, the SIF was centrifuged at 3000 g for 5 min to remove undissolved pancreatin fibers and bile. No loss in enzyme activity was observed upon centrifugation using a trypsin activity assay as described in the INFOGEST2 protocol (31) (data not shown). Just prior to the in vitro gastrointestinal digestion, a 0.3 M CaCl2 solution was added to simulated salivary, gastric and intestinal fluid in 1:200th, 1:2000th and 1:500th part respectively.
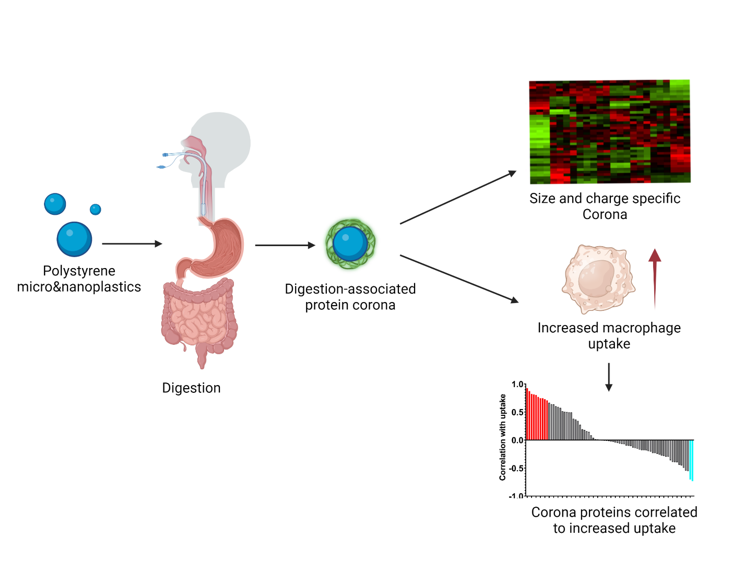
At the start of the digestion, 125 µl of a 2.5% MNP suspension (i.e., 3.125 mg total MNP mass) was sonicated in an ultrasonic bath (VWR, Amsterdam, Netherlands) to ensure a homogeneous suspension and was added to a 2 mL Eppendorf vial. First, 125 µl of 2X concentrated SSF was added to the MNPs in each sample, followed by manual mixing through inversion and a 5 min incubation at RT. Then, 250 µl of SGF was added to each sample, and the pH was adjusted to pH 3.0 ± 0.5 using 1 mM HCl. The samples were incubated at 37°C for 2 h while rotating head-over-heels at 25 RPM. Finally, 500 µl of SIF was added to each sample and the pH was adjusted to 7.0 ± 0.5 using 1 mM HCl, and the samples were incubated for 2 h at 37°C while rotating head-over-heels. For each experiment, a digestion blank including demi-water instead of MNP suspension was included. The digestion matrix needed to be diluted 25X in complete cell culture medium (CCM) before cell exposure to prevent matrix cytotoxicity (Supplementary Fig. S1).
To assess the protein corona formed upon in vitro gastrointestinal digestion or upon incubation (of nondigested) PS MNPs in CCM, PS MNPs were also incubated in CCM for 2 h while rotating head-over-heels at 37°C. We will refer to PS MNPs that were suspended in serum-free medium as pristine MNPs and those that were subjected to in vitro gastrointestinal digestion as digested MNPs. PS MNPs that were incubated in FCS-containing CCM will be referred to as serum-coated MNPs, and PS MNPs that underwent digestion and subsequent incubation in CCM will be referred to as digested+serum-coated MNPs (see Table 3). After digestion or incubation with serum, the samples were used immediately for MNP characterization or cell exposure.
Table 3: Nomenclature of MNPs according to their pretreatment and incubation in medium
|
|
Without in vitro intestinal digestion
|
Upon in vitro intestinal digestion
|
|
Medium without serum
|
Pristine MNPs
|
Digested MNPs
|
|
Medium with serum
|
Serum-coated MNPs
|
Digested+serum-coated MNPs
|
2.4. Characterization of particle suspensions
The hydrodynamic sizes of the PS MNPs before and after in vitro gastrointestinal digestion were determined by dynamic light scattering (DLS) using a ZS-nano zetasizer (Malvern Panalytical, Malvern, Great Britain). Cell culture medium without serum was filtered through a 0.2 µm nylon Whatman filter to remove background particulate matter. Pristine MNPs, digested+serum-coated MNPs and serum-coated MNPs were diluted to a concentration of 125 µg/ml in serum-free cell culture medium. The suspensions were sonicated for 1 min using a sonication bath and were subsequently loaded into a 1.5 mL polystyrene cuvette. The particle sizes were determined under a scattering angle of 173° and at a temperature of 25°C. At least 5 autocorrelation curves were obtained for each sample, and all particles were measured in triplicate. The remaining zeta-sizer settings were left at their default value. For each set of measurements, a medium blank was included to determine background particulate matter.
To determine potential fluorophore leaching from the MNPs upon in vitro gastrointestinal digestion, MNPs were removed from the gastrointestinal digestion supernatant using centrifugation at 30,000 g for 30 min, and the fluorescence intensity of the supernatant was measured using a Spectramax iD3 Multi-Mode Microplate Reader (Molecular Devices, Birkshire, United Kingdom). Leaching of fluorophores from the MNPs was not detected (Supplementary Fig. S2).
2.5. Assessing particle dosimetry
We assessed the fraction of MNPs deposited on the cells using the in vitro sedimentation, diffusion and dosimetry (ISDD) model (47). The ISDD model was run assuming a particle density of 1.05 g/ml, and assuming no agglomerate formation, the effect of protein binding was not considered for predicting particle sedimentation. The column height was set to 2.631 mm (corresponding to a 48-well plate format), and the medium volume was set to 0.5 ml. The grid was set to consist of 300 compartments, and sedimentation up to 24 h after cell exposure was simulated. The remaining settings were left at their default conditions. The predicted deposited dose is shown in the Supplementary materials (Supplementary Fig. S3).
2.6. Cell culture
THP-1 cells were obtained from ATCC (Manassas, USA) and were grown in RPMI 1640 medium (A10491, Thermo Fisher, Waltham, USA) supplemented with 10% fetal calf serum and 1% penicillin/streptomycin. The cells were cultured in a humidified incubator at 37°C at 5% CO2. Cells were maintained at a cell density between 2*105 and 8*105 cells/ml in an upright T75 grainer culture flask and were passaged twice a week. THP-1 cells were seeded at 5*105 cells/ml in all experiments. For cell viability experiments, 100 µl THP-1 cells were seeded in 96-well plates and subsequently differentiated to M0 macrophages by the addition of 20 ng/ml phorbol 12-myristate 13-acetate (PMA) and were incubated for 48 h in a humidified incubator (48). After that, the PMA-containing medium was replaced with PMA-free complete cell culture medium, and the cells were allowed to rest for 24 h. On the subsequent day, the cells were exposed to MNPs or blanks for up to 24 h.
2.7. Cell viability assessment
To derive noncytotoxic concentrations of the gastrointestinal digestion matrix and MNPs to THP-1-derived macrophages, a water-soluble tetrazolium dye 1 (WST-1) assay was performed. Briefly, THP-1 cells were seeded and differentiated to macrophages in 96-well plates as described above (see section 2.6). Only nonfluorescent MNPs were used for the cell viability assay to prevent potential interference of the fluorescent label with the WST-1 viability assay. Based on the cytotoxicity of the digestion matrix (Supplementary Fig. S1), the digested MNPs and serum-coated MNPs were diluted 1:25 with complete culture medium to a concentration of 125 µg/ml. THP-1-derived macrophages were exposed to digested+serum-coated-MNPs or serum-coated-MNPs at concentrations of 7.8, 15.6, 31.3, 62.5, and 125 µg/ml for 24 h. Next, the medium was aspirated, and the cells were washed with PBS to remove MNPs that were not associated with the cells. Then, 100 µl of culture medium containing 5% WST-1 reagent was added to each well, and the plates were incubated for 1 h at 37°C while shaking at 300 RPM. After 1 h, the absorbance at 440 nm was measured using a Spectramax iD3 Multi-Mode Microplate Reader. The MNPs showed no signal in a cell-free WST-1 assay (data not shown), excluding potential particle interference.
For each experiment, a positive control containing 0.5% Triton X-100 and a negative control consisting of complete culture medium were included. The viability was calculated by comparing the absorbance at 440 nm to the negative control and expressing the resulting absorbance as a percentage of the negative control. The data were analyzed using GraphPad Prism 9.3.1 (GraphPad Software, LLC, San Diego, CA, USA), and a two-way ANOVA using Dunnett’s post hoc correction was performed to assess significant differences from the medium control. Experiments were performed in triplicate.
2.8. Quantitative measurements of cell association
MNP cell association was measured using flow cytometry. Briefly, THP-1 cells were seeded in a 48-well Nunclon plate (Thermo Fisher) and were differentiated to macrophages as described above (see section 2.6). The THP-1-derived macrophages were exposed to digested+serum-coated or serum-coated MNPs for up to 24 h. The cells were subsequently fixed using 4% paraformaldehyde in PBS for 15 minutes at room temperature and washed twice using PBS. After fixation, the cells were stored at 4°C until measurement using flow cytometry for at most 24 h. Prior to flow cytometry, 1 mM EDTA was added to the cell suspension, and the cells were detached from the bottom of the well using a curved Pasteur pipette and transferred to a 96-well Nunclon plate. Cell fluorescence was measured in triplicate, each consisting of 3 technical replicates, using a Cytoflex LX flow cytometer in plate mode (Beckman Coulter NL, Woerden, Netherlands). MNP cell association was assessed based on the fluorescein isothiocyanate (FITC) channel (525 nm:20), which corresponds to the green fluorescence emitted by the MNPs. Cells were distinguished from cell debris based on their forward and side scatter, and at least 5000 cells were analyzed for each of the samples. Cells that had a FITC signal higher than 99% of the cells in the negative control were considered FITC positive. The cell association of MNPs over time of two different concentrations of MNPs (i.e., 15.6 and 62.5 µg/ml) was analyzed to determine the optimal measurement time (Supplementary Fig. S4 & S5). Next, the cell association upon 24 h incubation with 7.8, 15.6, 31.3, 62.5 or 125 µg MNPs/ml was assessed (n=3). To correct for differences in fluorescence intensity between the different MNPs, the obtained relative light unit (RLU) values were divided by the relative fluorescence intensity of each of the MNPs at 525 nm as determined using a luminometer (Supplementary Fig. S6). Then, the corrected fluorescence was expressed as the fold change compared to the average fluorescence intensity of unexposed cells.
2.9. Determination of cellular internalization
Internalization of fluorescent MNPs into THP-1-derived macrophages was assessed using a Rescan fluorescence confocal microscope (Amsterdam, The Netherlands). First, THP-1 cells were seeded and differentiated into macrophages as described above (section 2.6) in 8-well µ-slides with a tissue culture-treated glass bottom designed for confocal imaging (Ibidi GmbH, Gräfelfing, Germany). The cells were subsequently exposed to 125 µg/ml of serum-coated or digested+serum-coated MNPs. A digestion blank and negative control containing only culture medium and cells were also included. After 1 h of MNP exposure, the cells were washed once with 300 µl of PBS, and the cell surface was stained with 10 ng/ml wheat-germ agglutinin in blocking buffer (2% FBS in PBS) for 1 h. The cells were fixed and washed as described in section 2.8.
2.10. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis
The composition of the protein corona was qualitatively assessed using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‒PAGE), based on the protocol published by Walczak et al. 2015 (24). Digested, digested+serum-coated and serum-coated MNPs were separated from the unbound proteins by centrifugation at 30,000 g for 30 min using a tabletop centrifuge. The supernatant was discarded, and the pellet was washed with 1 mL of PBS followed by manual resuspension through pipetting and 10 seconds of vortexing followed by another centrifugation. The washing step and subsequent centrifugation were repeated twice. Next, the pellet was resuspended in 100 µl of 1X Laemmli sample buffer using a sonication bath followed by vortexing for 10 seconds, and the samples were subsequently boiled at 95 °C for 5 min. To reduce the amount of MNPs loaded into the SDS‒PAGE gel, MNPs were separated from the protein fraction dissolved in Laemmli buffer by centrifugation at 30,000 g for 5 min. An equal volume of protein of each MNP sample was loaded into a SurePAGE MOPS-Tris 10-well PAGE gel (Genscript EU, Netherlands), and the gel was run at 120 V. Afterwards, the gels were washed twice with demi-water for 10 min to remove excess SDS. The gels were submerged into fixation solution (10% glacial acetic acid, 40% methanol, 50% demi-water) for 20 min followed by a washing step with demi-water for 10 min. The gels were stained using Biosafe G-250 Coomassie for 2 h and were destained with water overnight. An Odyssey gel-imaging system (Li-COR, Homburg vor der Höhe, Germany) was used to capture the fluorescence at 700 nm originating from Coomassie blue, and subsequent images were analyzed using Fiji 2.9.0 as previously published (49).
2.11. Semiquantitative protein corona determination by proteomics
The composition of the protein corona was quantified after in vitro gastrointestinal digestion, serum incubation or the combination using an on-bead protein digestion protocol based on the protocol described by Wendrich et al. (2017) (50). After in vitro gastrointestinal digestion or incubation in culture medium, the MNPs were centrifuged at 30,000 g for 30 min. The supernatant was discarded, and the pellet was washed with 1 mL of 1 M ammonium bicarbonate buffer followed by 5 seconds of sonication and 10 seconds of vortexing. The washing step was repeated once with 1 M ammonium bicarbonate buffer, and afterwards, the pellet was washed with 50 mM of ammonium bicarbonate buffer. The supernatant was removed, and the MNPs were resuspended in 50 µl of 50 mM ammonium bicarbonate buffer. The protein sample was chemically reduced by adding 5 µl of freshly prepared 150 mM dithiothreitol and was incubated for 30 min at 45°C while shaking at 500 RPM. Five microliters of 200 mM iodoacetamide was added, and the sample was incubated at 20°C in the dark for 30 min. Six microliters of 200 mM of cysteine in ammonium bicarbonate buffer was added to stop the alkylation by iodoacetamide, followed by the addition of 500 ng of sequencing grade trypsin for an overnight digestion at 25°C while shaking at 350 RPM. The following day, the enzymatic digestion was stopped by the addition of 3 µl of 10% tri-fluoro acetic acid to the samples. The peptide samples were cleaned using the µ-column method as previously described (51). The solvent remaining after µ-column sample clean-up was removed using a rotary evaporator, and the peptide samples were redissolved in 50 µl of 1% formic acid in demi water. The samples were subsequently measured using a nano LC‒MS-MS protocol as previously described (23). Briefly, 5 µl of tryptic peptide solution was injected into a 0.10 × 250 mm ReproSil-Pur 120 C18-AQ 1.9 μm beads analytical column (prepared in-house) at 800 bar. A gradient from 9 to 34% acetonitrile in water with 0.1% formic acid in 50 min (Thermo Vanquish Neo) was used. Full scan FTMS spectra were obtained using an Orbitrap Exploris 480 Thermo electron (San Jose, CA, USA) in positive mode between 380 and 1400 m/z. The 25 most abundant positively charged peaks in the MS scan were fragmented (HCD) with an isolation width of 1.2 m/z and 24% normalized collision energy. Samples were measured in triplicate. Since the in vitro digestion protocol contains proteases in addition to trypsin, nonspecific digestion was assumed. To account for peptides originating from digestion by non-trypsin proteases, an in-house database of nonspecific porcine and bovine peptides was generated. Proteins in each sample were identified by comparing the identified peptides to the in-house bovine and porcine databases.
2.12. Analysis of LC‒MS-MS-based proteomics data
The label-free quantification (LFQ) values were obtained from MaxQuant (52) and were analyzed using Perseus (53). First non-bovine or porcine contaminant proteins, proteins that did not have at least 2 valid values in any condition and proteins only identified by modified peptides (only identified by site) were removed from the dataset. Then, protein annotations, including GO biological process, molecular function, protein family and KEGG function, were retrieved using the Bos Taurus and Sus Scrofa UniProt databases (54). To visualize proteins present on the PS MNPs upon different treatment conditions, a Venn diagram and upset plot were generated in R using the R Graph gallery VennDiagram (55) and the UpSetR (56) packages. The raw LFQ values were log2 transformed, and missing values were imputed from a normal distribution using a downshift of 3.0 and default band narrowing. The log2 transformed values were used to generate a PCA plot, a heatmap of the Pearson correlation coefficient and a hierarchical clustering of the protein abundance. Differentially abundant proteins were identified using a two-tailed T test with permutation-based false discovery rate (FDR) correction using 250 simulations. Proteins were considered differentially abundant if they had a p value smaller than 0.01 and an absolute log2-fold change larger than 2. The results were plotted as volcano plots using the volcano plot function of Perseus. The correlation between particle uptake and protein abundance was computed in GraphPad 9 by taking the luminescence values obtained from flow cytometry for cells treated with the highest concentration of MNPs and by subsequently using the GraphPad 9 correlation function to correlate this to the averaged protein LFQ value obtained for each of the treatment-particle combinations. Proteins that showed a Pearson correlation coefficient larger than 0.5 were considered correlated, and the resulting correlated proteins were manually compared to previous publications (57, 58) and the annotations obtained from the UniProt database (54). To assess enrichment of protein functions in differentially abundant proteins and proteins correlated with uptake, a Fischers exact test with Benjamini‒Hochberg-based FDR correction was performed to determine if there was a significant association between the proteins of interest and specific GO terms. KEGG and GO functions with an FDR smaller than 0.05 were considered significantly enriched.
{kind=link}