MFMs are rare, inherited or sporadic, progressive and genetically heterogeneous disorders.[7]
They are defined morphologically by the presence of foci of myofibril dissolution
that begin at the Z-disk, an accumulation of myofibrillar degradation products, and
the ectopic expression of a large number of proteins including desmin, dystrophin,
and ubiquitin. MFM subtypes are designated according to the affected protein, such
as desminopathy, aB-crystallinopathy, Bag3opathy, or filaminopathy.[8] MFM subgroup
frequencies vary depending on the cohort, with filaminopathy accounting for around
4% of all MFM patients according to the Mayo clinic cohort.[9]
Filaminopathy is caused by FLNC mutations. Patients with mutations in the FLNC Ig-like domain usually present with weakness in limb-girdle muscles, and typical
histological characteristics include focal disintegration of myofibrils and the aggregation
of sarcoplasmic proteins in muscle biopsies.[3,10] Another MFM type is caused by missense
mutations in the actin binding domain of FLNC, which leads to increased actin binding affinity. These patients were described as
having a distal myopathy without typical pathological features of MFMs. A frameshift
mutation in the Ig-like domain, resulting in haploinsufficiency in FLNC, was also shown to be causative of distal myopathy.[11-12]
Patients presenting with distal filaminopathy usually onset at the age of 30–40
years. The most common onset symptom is weakness of the upper limbs, with distal extensors
typically more severely affected than flexor muscles. Atrophy is seen mostly in interosseus
muscles, especially the first dorsal, and asymmetry is noted in about one-third of
patients.[13-14] Our patient shared the same characteristics. A previously reported
Australian family including nine affected individuals with distal filaminopathy showed
slow progression of muscle weakness. Lower extremities were affected around 10 years
after disease onset. Progression to the proximal muscles was evident in the fifth
decade of life and patients usually required a stick for walking during their sixth
decade.[11] Our patient showed a more severe clinical pattern. His lower extremities
were affected around 6 months after disease onset, and his ambulation is currently
limited. This may have been caused not only by muscular lesions, but also by the involvement
of peripheral nerves and motor neurons in the spinal cord that presented as LMN syndromes.
LMN syndromes are clinically characterized by muscle atrophy, weakness, and hyporeflexia
without sensory involvement. They may arise from disease processes affecting the anterior
horn cell or the motor axon and/or its surrounding myelin. Neurophysiological analysis
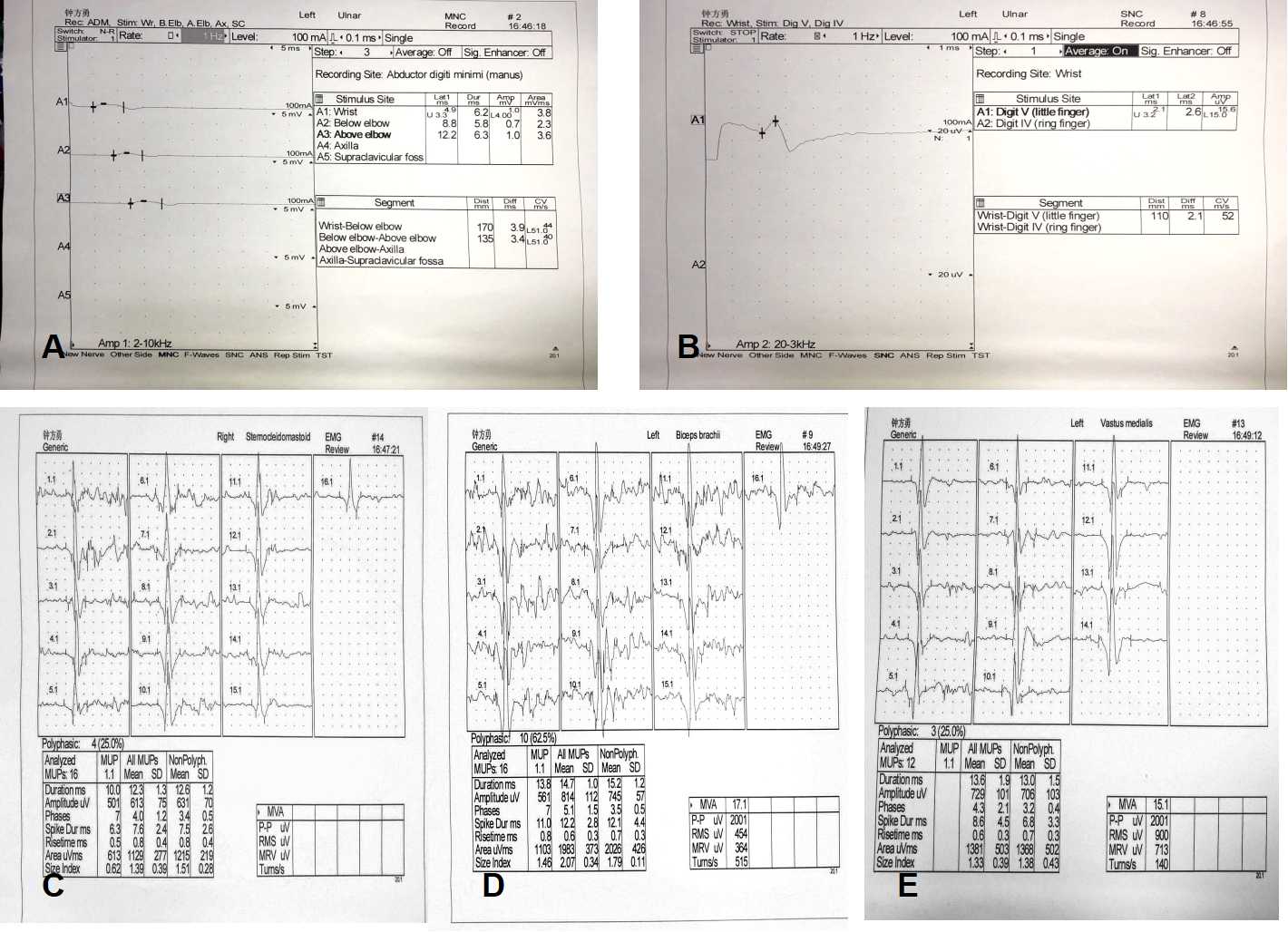
may help support their diagnosis.[7,15] Our patient’s nerve conduction velocity revealed
obviously diminished CMAP amplitudes in the motor nerves, and absent right ulnar nerve
F-waves. The presence of chronic denervation was confirmed by long duration, large
amplitude, and decreased motor unit recruitment on the EMG. This was observed in the
cervical, thoracic, and lumbosacral spinal cord regions, which may indicate that the
roots and branches of peripheral nerves, even motor neurons, were damaged. Such neurophysiological
findings have not been reported in previous cases of distal filaminopathy, which mainly
showed decreased CMAP amplitudes and a myogenic pattern on EMG. Only one patient with
distal filaminopathy had mixed myogenic and neurogenic changes in the anterior tibial
muscle.[11,13-14] While obvious neuropathic changes on EMG can be observed in MFM
patients, genetic testing is nevertheless important to help make a correct diagnosis.
LMN syndromes can be hereditary, sporadic, or immune-mediated, and include multifocal
motor neuropathy, chronic inflammatory demyelinating polyneuropathy, SMAs, Kennedy’s
disease, distal hereditary motor neuropathies, and motor neuron disease.[6,15] To
carry out a differential diagnosis in our patient, we tested for the presence of peripheral
neuropathy antibodies, SMA genes, and the androgen receptor gene, but all were negative.
Therefore, we suggest that his LMN syndrome is associated with a FLNC mutation.
Peripheral nerve involvement has been reported in some MFM patients, including those
with desminopathy and ZASPopathy. Moreover, axonal spheroidal formations in the spinal
cord and spinal roots that were immunoreactive for neurofilaments were detected in
a postmortem of a desminopathy patient.[1,16] In a German filaminopathy cohort, a
muscle biopsy of a patient with hyperesthesia indicated neurogenic changes although
nerve conduction velocities were normal.[4] From our current investigation, we suggest
that the peripheral nervous system and even the spinal cord could be involved in filaminopathy,
with some patients showing a more severe clinical pattern. However, details of the
pathological changes and underlying mechanism of filaminopathy should be confirmed
by additional studies such as nerve biopsies.
The MRI of our patient disclosed a markedly increased signal intensity in the bilateral
gastrocnemius muscles in TIRM sequences, while only a minor increase was seen in T2
sequences. This suggested that the affected muscles had edema, and that they had deteriorated
rapidly. Patients in other studies with longer term courses and slower progress had
markedly increased signal intensities in T2, consistent with fat replacement and lack
of hyperintensity on TIRM sequences indicating an absence of muscular edema. The distribution
of affected muscles in our patient was typical. Muscle imaging of filaminopathy often
shows a homogenous pattern of muscle involvement. In patients with proximal muscle
weakness, the involvement of the vastus intermedius and medialis, adductor magnus,
and semimembranosus and biceps femoris muscles is more pronounced. The tibialis anterior
muscles, as well as the gastrocnemius and soleus muscles, are usually involved in
patients with distal muscle weakness.[3-4,10-13,17]
FLNC is located on chromosome 7q32-q35 and contains 48 coding exons. Mutation c.7123G>A,
p.V2375I in our patient is in the 21st Ig-like domain. Most patients with mutations
in FLNC Ig-like domains present with weakness in their proximal muscles and have typical
protein aggregation in muscle biopsies, while those with mutations in actin-binding
domains present with distal myopathy without typical pathological changes on histopathological examination.[13-14] However, our patient showed the opposite presentation.
Despite carrying a mutation in the Ig-like domain, he showed distal myopathy and had
typical protein aggregation on muscle biopsy with the presence of large inclusions in the muscle fibers. This suggests that mutations
in actin-binding and Ig-like domains cause overlapping clinical symptoms and histopathological
changes, but the relationship between genotype and phenotype remains unclear and may
be affected by as yet unknown genetic and environmental causes.[12]
In conclusion, we report a middle-aged Chinese male with a heterozygous missense
mutation (c.7123G>A, p.V2375I) in the Ig-like domain 21 of FLNC. He showed distal myopathy and lower motor neuron syndrome with a more severe clinical
pattern than other reported cases. Obvious muscle edema could be observed on MRI,
and typical protein aggregation presented in the pathological study. Our study not
only expands the clinical spectrum of filaminopathies, but also extends the histopathologic
and genetic heterogeneity of hereditary distal myopathies and lower motor neuron syndromes.
Nevertheless, underlying mechanisms and the relationship between the phenotype and
genotype need further study.
{kind=link}