Cell culture and materials
Human multiple myeloma cell lines NCI-H929, RPMI-8226, and human Burkitt’s lymphoma cell line Raji that were preserved in our laboratory were cultured in RPMI-1640 medium (Gibco, USA) that contained 10% (v/v) fetal bovine serum (FBS, Gibco, USA). Human natural killer cell line NK-92MI from our lab was cultured in Alpha Minimum Essential Medium (MEM-α, Gibco, USA) with 2 mM L-glutamine, 1.5 g/L sodium bicarbonate, 0.2 mM inositol, 0.1 mM 2-mercaptoethanol, 0.02 mM folic acid, 12.5% (v/v) horse serum (Hyclone), and 12.5% (v/v) FBS. Human embryonic kidney 293 (HEK 293) cells from our lab were cultured in Dulbecco's Modified Eagle Medium (DMEM, Gibco, USA) with 10% (v/v) FBS. All cells were incubated in a humid atmosphere of 5% CO2 at 37℃. The 4-w-old Balb/c nude mice used in this study were purchased from Comparative Medicine Centre of Yangzhou University (Yangzhou, China). All animals were fed in a specific pathogen-free environment and were treated according to the criteria of the Comparative Medicine Centre of Yangzhou University.
Construction, expression, purification, and identification of 2A9-MICA and 2A9-MICAα1-2
The single chain antibody 2A9 that targeted human BCMA was screened from the CPU2 phage display library of murine single-chain antibodies using phage display technology (Wang et al. 2020). The cDNA sequences of 2A9 and human MICA extracellular domains (residues 24–307, Uniprot identifier: Q29983) were optimized to HEK 293-preferred codons at GenScript (Nanjing, China). Then, we spliced 2A9 with hMICA and 2A9 with hMICAα1-2 by overlap PCR after digestion with EcoR Ⅰ and Not Ⅰ (Takara, Japan). The recombinant DNA of 2A9, 2A9-MICA, and 2A9-MICAα1-2 were ligated separately to a digested pcDNA 3.0 vector, which formed pcDNA 3.0-2A9, pcDNA 3.0-2A9-MICA, and pcDNA 3.0-2A9-MICAα1-2. Further, these three recombinant eukaryotic expression vectors were transfected into HEK 293 cells with polyethylenimine (PEI). During the culture of transfected host cells, the culture supernatants were collected to purify proteins 2A9, 2A9-MICA, and 2A9-MICAα1-2 by affinity chromatography on a His Trap high-performance column (GE Healthcare, Sweden), which was followed by analysis using SDS-PAGE (15% gel for 2A9; 10% gel for 2A9-MICA and 2A9-MICAα1-2) and Western blot assay (anti-His antibody for 2A9, 2A9-MICA, and 2A9-MICAα1-2, Proteintech Group, Wuhan, China; anti-MICA antibody for 2A9-MICA and 2A9-MICAα1-2, SunBio Technology, Nanjing, China).
Quantitative real-time polymerase chain reaction (qRT-PCR)
RNAs were isolated using TRIeasy™ Total RNA Extraction Reagent (Yeasen, Shanghai, China) and transcribed into cDNA using Hifair® Ⅱ 1st Standard cDNA Synthesis Kit (Yeasen, Shanghai, China). Gene transcripts were detected using an Applied Biosystems 7300 Plus Real-Time PCR System (Thermo Fisher Scientific, USA) and Hieff UNICON® qPCR SYBR Green Master Mix (High Rox, Yeasen, Shanghai, China) and gene-specific primers (BCMA up: 5’ACCTTGTCAACTTCGATGTTCTT3’, BCMA down: 5’CAGAGAATCGCATTCGTTCCTT3’; GAPDH up: 5’TGTGGGCATCAATGGATTTGG3’, GAPDH down: 5’ACACCATGTATTCCGGGTCAAT3’). The GAPDH internal control was used for normalization. Average fold-change values of gene expression were calculated from triplicate measurements using the ∆∆CT method.
Immunoblot
Immunoblots were performed according to standard procedures using anti-human-BCMA (SunBio Technology, Nanjing, China) and anti-GAPDH (Proteintech Group, Wuhan, Chima) antibodies and HRP-conjugated secondary reagents (goat-anti-rabbit IgG and goat-anti-mouse IgG, all from Proteintech Group).
Analysis of binding affinity using flow cytometry and surface plasmon resonance
To evaluate the binding affinity of the fusion proteins to BCMA+ multiple myeloma cells, NCI-H929 and RPMI-8226 were used as experimental objectives and Raji was the negative control; 1×106 cells of each were pre-incubated with PBS that contained 2% (v/v) FBS and 1000 nM protein samples or an isotype control antibody at 4℃ for 1 h. Cells were then incubated with mouse His-tag monoclonal antibody and CoraLite488-conjugated Affinipure goat anti-mouse IgG (H+L) (Proteintech Group, Wuhan, China) for 1 h at 4℃. The cells were analyzed by flow cytometry (MACSQuant, Miltenyi Biotec, Germany).
The binding kinetics of fusion proteins 2A9-MICA and 2A9-MICAα1-2 to the NKG2D protein were measured with a Biacore system (Biacore X100, GE Healthcare, Sweden). Human NKG2D protein (Novoprotein, Shanghai, China) was coupled covalently to the CM5 sensor chip (GE Healthcare, Sweden), and soluble 2A9-MICA or 2A9-MICAα1-2 was injected at gradient concentrations into the running buffer (HBS-EP, pH 7.4). One flow cell of the sensor chip was used as a control, another flow cell was the experimental channel, and Gly-HCl (pH 2.0) was used to elute the analyte. The affinity of fusion proteins with NKG2D was evaluated with Biacore X100 Evaluation software: the equilibrium dissociation constant (KD) = dissociation rate constant (kd) / association rate constant (ka).
NKG2D expression on NK-92MI and cytotoxicity assay
NK-92MI cells were stained with PE-anti-human CD314 (NKG2D) antibody (Biolegend, USA) and analyzed by flow cytometry to determine NKG2D expression. A Lactate Dehydrogenase (LDH) Cytotoxicity Assay Kit (Beyotime, Shanghai, China) was used to detect LDH release from target cells in a cytotoxicity assay. The target cells NCI-H929, RPMI-8226, and Raji were co-cultured with various amounts of NK-92MI cells for 6 h at 37℃ in the presence of different treatments; 60 μL of the co-culture supernatants from each well was then analyzed for LDH activity. In addition to the experimental groups, a spontaneous LDH release in effector or target cells and a maximum LDH release in target cells were also prepared. Absorbance values were used to calculate the rate of cell lysis. Cell lysis (%) = [(experimental-effector cells spontaneous-target cells spontaneous)/(target cells maximum-target cells spontaneous)]×100%.
Stability of fusion proteins
2A9, 29-MICA, and 2A9-MICAα1-2 (2 mg/mL) were stored at 4℃ and -20℃ for 5 d, and then SDS-PAGE was performed to detect the degradation of proteins under different short-storage conditions to evaluate their physical stability. 2A9, 2A9-MICA, and 2A9-MICAα1-2 were diluted with filtered human plasma to 1000 nM and kept at 37°C for 1, 3, 5, or 7 d. Flow cytometry was used to detect the binding of proteins in human plasma at 37℃ to NCI-H929 cells for a preliminary analysis of their biological stability.
NCI-H929 xenograft model
The NCI-H929 cells (1x107) were injected subcutaneously into the right dorsal flank of each 4-w-old Balb/c nude mouse (Hipp et al. 2017; Seckinger et al. 2017). When the average tumor volume reached 100 mm3, all the tumor-bearing mice were divided randomly into four groups of five mice each; fusion proteins or other treatments were administered to each group according to established protocols. The anti-myeloma efficacy of fusion proteins 2A9-MICA and 2A9-MICAα1-2 (i.v., 2 mg/kg, q.2d.) was estimated compared with Rd treatment (i.g.; R: Lenalidomide, 3 mg/kg, q.d.; d: Dexamethasone, 5 mg/kg, d1-4/d9-12), and the saline-treated mice were set as the vehicle control. During the administration, 5×106 NK-92MI cells were injected intravenously into every experimental mouse (q.6d.) (Swift et al. 2012; Zhu et al. 2017). Tumor growth and body weight of mice were monitored by caliper measurements every other day (i.e., Tumor volume: V=LW2/2, L: the longest diameter of tumor, W: maximum transverse diameter of vertical direction). The study was terminated on day 22 after administration when tumors of vehicle-treated mice exceeded 2000 mm3. Tumor tissues and organs (i.e., heart, liver, spleen, lung, and kidney) of mice were prepared for further analysis.
Immunohistochemistry, immunofluorescence, and hematoxylin-eosin (H&E) staining
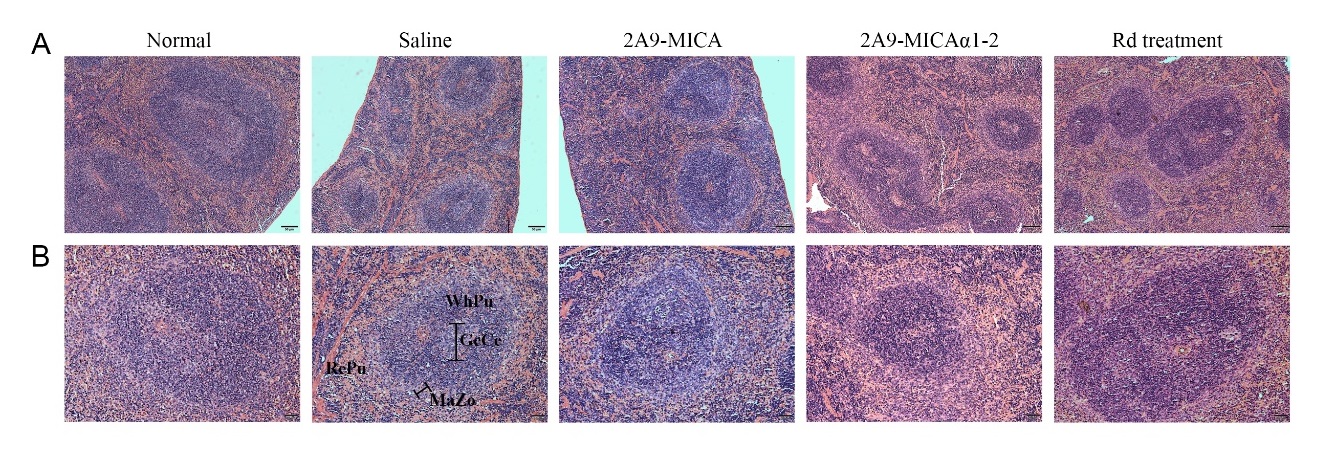
Tumor tissues from experimental mice and spleens of normal and experimental mice were fixed by 4% paraformaldehyde and embedded in paraffin, and paraffin blocks were cut into 2 μm sections. For immunohistochemical (IHC) staining, tumor sections were incubated with anti-Ki-67 antibody (Cell Signaling Technology, Boston, USA) and anti-BCMA antibody (Proteintech Group, Wuhan, China) separately, followed by HRP-conjugated goat anti-rabbit secondary antibody, then detected with DAB Immunohistochemistry Color Development Kit (Sangon Biotech, Shanghai, China). For immunofluorescence (IF) staining, tumor sections were incubated with anti-human CD56, anti-TNF-α antibodies (Novus Biological, USA), and Alexa Fluor® 647 AffiniPure Goat Anti-Rabbit IgG (H+L) (FMS-Rbaf64701, FcMACS, NanJing, China). Spleen sections were used for H&E staining to evaluate the splenic damage and toxicity of fusion proteins. Along with these assays, images of IHC and H&E staining sections were captured with an upright biological microscope (BX53, Olympus, Japan) or inverted fluorescence microscope (ECLIPSE Ts2R, Nikon, Japan). Fluorescent images were observed with a confocal laser scanning microscope (CLSM, LSM800, Zeiss, Germany) and processed using ZEN imaging software.
Statistical Analysis
Data were analyzed using SPSS 22.0 and GraphPad Prism 6. Differences between multiple groups were estimated by a Student t test, and a P-value < 0.05 was considered to be statistically significant.
{kind=link}
{kind=link}
{kind=link}
{kind=link}