Cells and reagents
LentiX-293T cells were purchased from Takara (Shiga, Japan). Plat-E cells were a generous gift from Dr. Kitamura (The University of Tokyo, Tokyo, Japan). Osteoblasts and limb bud cells were cultured in alpha modification of Eagle’s minimum essential media (α-MEM; Thermo Fisher, Waltham, MA, USA) containing 10% fetal bovine serum (FBS) at 37 °C in a humidified 5% CO2 incubator. Plat-E cells and LentiX-293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Thermo Fisher) containing 10% FBS. Recombinant Bmp2 was obtained from conditioned medium of LentiX-293T cells transfected with a Bmp2 expression vector as described previously (41). Bmp2 activity was determined by comparison with human recombinant Bmp2 (Peprotech, Rocky Hill, NJ, USA).
Osteoblasts were isolated from calvariae of 3–5-day-old neonatal mice by a sequential enzymatic digestion method as described previously (42). Briefly, mouse calvariae were gently incubated with 5 mM ethylenediaminetetraacetic acid (EDTA) in phosphate-buffered saline (PBS) for 1 h at 37 °C, followed by three 20-min digestions with 0.25% collagenase in DMEM for 20 min at 37 °C. Cells obtained during the last two digestion processes were collected together in α-MEM containing 10% FBS. Throughout the subsequent experiments, α-MEM containing 10% FBS, 50 μg/mL ascorbic acid, and 5 mM sodium β-glycerophosphate was used to induce osteoblastic differentiation. Limb bud cells were isolated from mouse embryos at E12–E13 and digested with 0.05% trypsin/0.53 mM EDTA in PBS for 10 min at 37 °C. Cells obtained during the digestion were collected in α-MEM containing 10% FBS. For monolayer culture, limb bud cells were seeded at 1.6 × 105 cells/cm2. All other chemicals used were of the highest purity commercially available.
RNA-sequence and data analysis
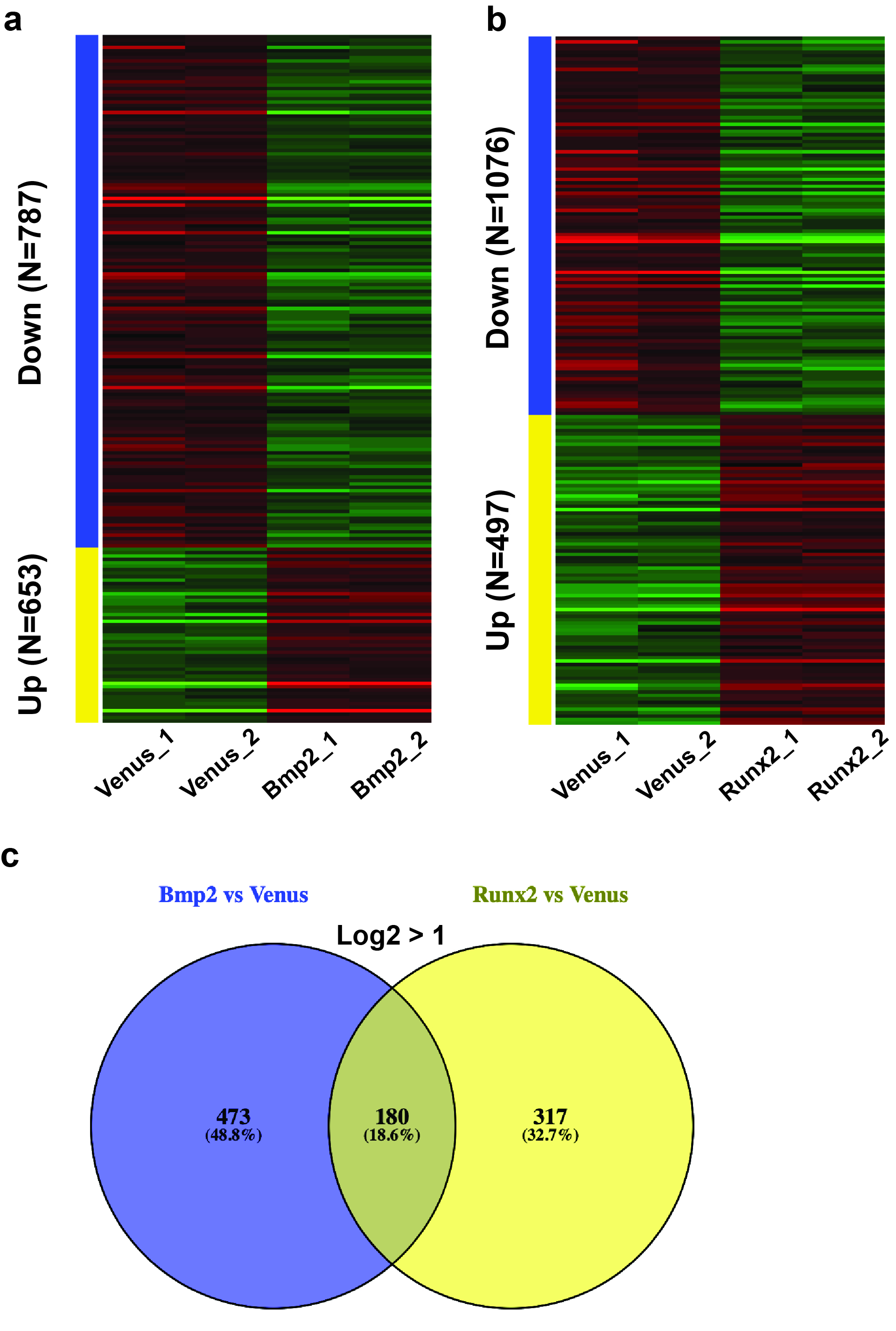
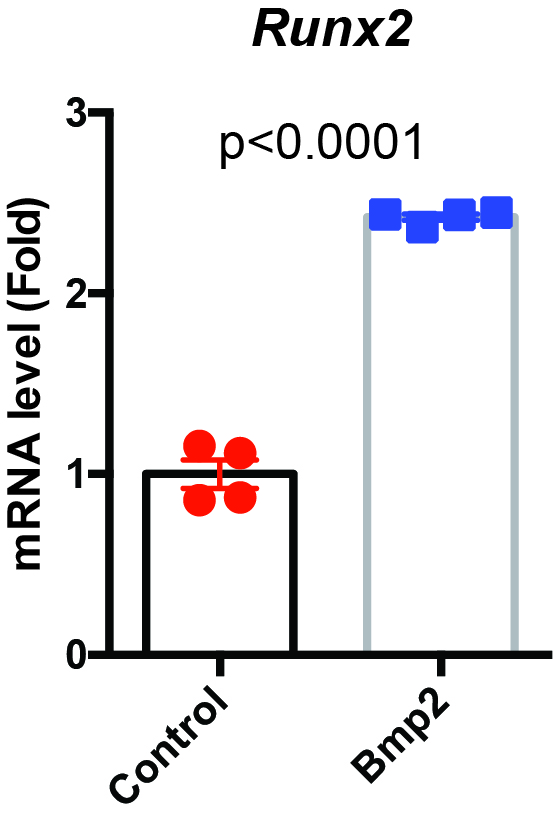
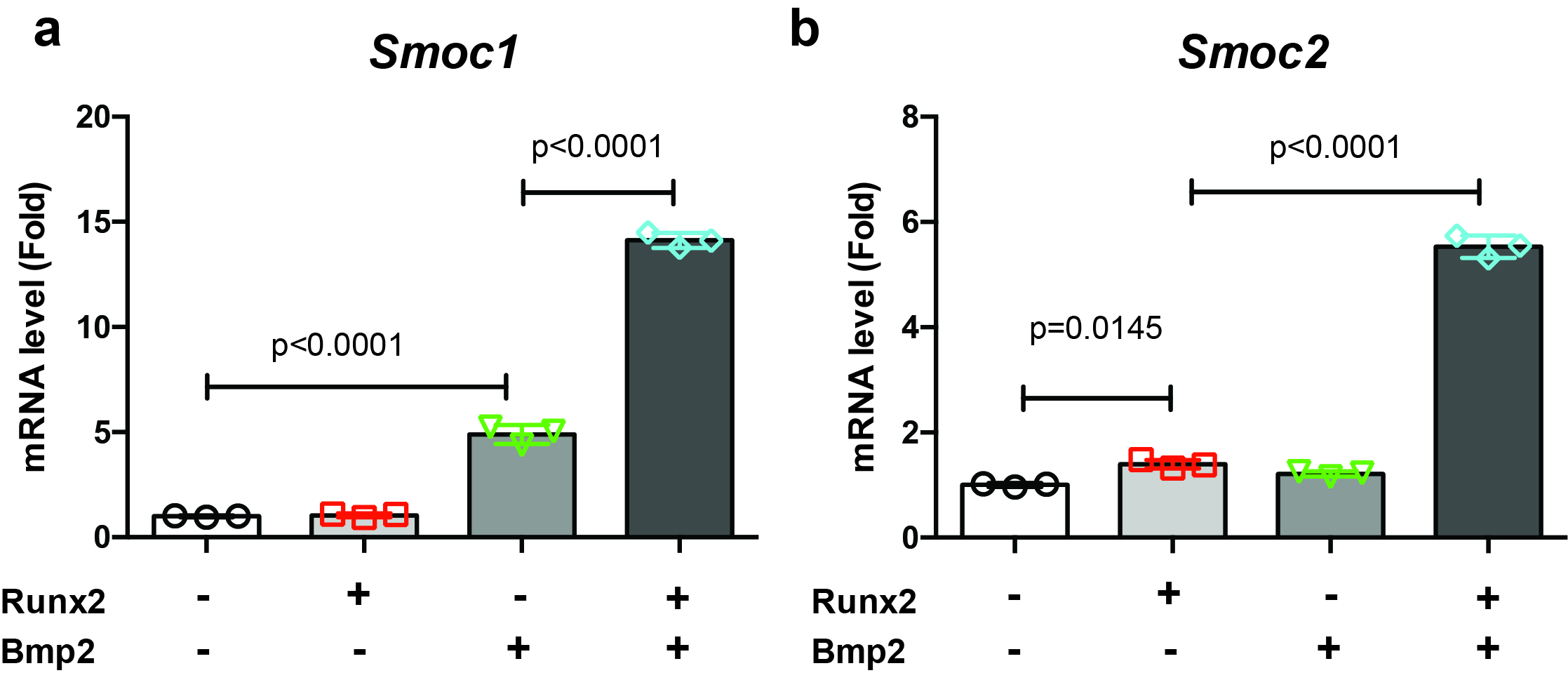
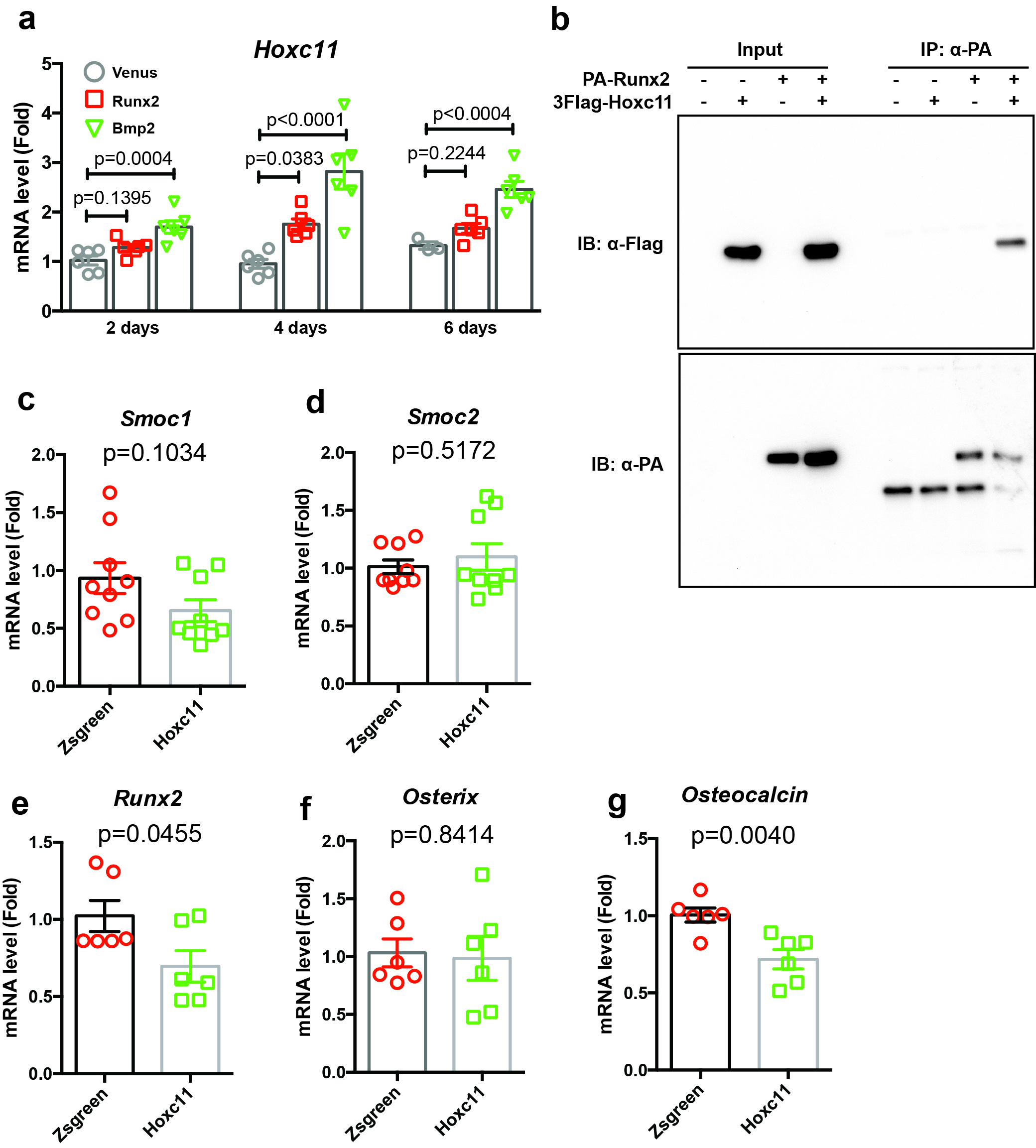
Limb bud cells were infected with Venus, Runx2, or Bmp2 adenovirus. After 4 days of incubation, total RNA was extracted using a Nucleospin RNA Plus Kit (Takara). Sequencing was performed on an Illumina HiSeq 2500 platform (Illumina, San Diego, CA, USA) in a 75-base single-end mode. Sequenced reads were mapped to the mouse reference genome sequences (mm10) using TopHat version 2.1.1 in combination with Bowtie 2 version 2.4.2. and SAMtools version 1.11. The fragments per kilobase of exon per million mapped fragments were calculated using Cufflinks version 2.2.1. RNA-sequencing data was analyzed by iDEP 9.1. Raw reads from these samples were submitted to the National Center for Biotechnology Information Gene Expression Omnibus database (accession number: GSE166982).
Plasmids
Venus, Runx2, DN-Runx2, or Bmp2 cDNAs were ligated to the pAXcawt adenovirus vector (Takara) as described previously (43). Flag-tagged-DN-Runx2 used in this study contains amino acids 2–247 of Runx2 (11). This construct lacks the transcriptional activation domain at the C-terminal region. The generation of these adenoviruses was performed using an adenovirus generation kit (Takara). A Venus adenovirus was used as the control adenovirus (44). For shRNA vector construction for Smoc1 and Smoc2, the following oligo DNAs were used:
shSmoc1 Forward, 5′-GATCCGCAAAGACTCCAAGTTGAATAATTCAAGAGATTATTCAACTTGGAGTCTTTGTTTTTTG-3′ and Reverse, 5′-AATTCAAAAAACAAAGACTCCAAGTTGAATAATCTCTTGAATTATTCAACTTGGAGTCTTTGCG-3′; shSmoc2 Forward, 5′-GATCCGCCAAGAATGACAATGTAGTGATTCAAGAGATCACTACATTGTCATTCTTGGTTTTTTG-3′ and Reverse, 5′-AATTCAAAAAACCAAGAATGACAATGTAGTGATCTCTTGAATCACTACATTGTCATTCTTGGCG-3′; shGFP control Forward, 5′-GATCCGCACAAGCTGGAGTACAACTACTTCAAGAGAGTAGTTGTACTCCAGCTTGTGTTTTTTG-3′ and Reverse, 5′-AATTCAAAAAACACAAGCTGGAGTACAACTACTCTCTTGAAGTAGTTGTACTCCAGCTTGTGCG-3′. Each pair of single-stranded oligo DNAs was annealed at a concentration of 25 μM, and incubated at 95 °C for 5 min. The annealed oligo DNAs were individually inserted into the pSIREN-retroQ-based shRNA expression vector (Takara) at the BamHI/EcoRI site.
Retrovirus infection
PLAT-E cells were seeded at 8 × 104 cells/cm2 at 1 day before transfection. Polyethylenimine (PEI) was used for all transfections. The pSIREN-retroQ shRNA expression vectors for shSmoc1 and shSmoc2 were mixed with PEI, and the plasmid-PEI complexes were incubated in Opti-MEM (Thermo Fisher) for 15 min at room temperature, and added to PLAT-E cells. The virus supernatant was collected at 48 h after transfection, and used to infect osteoblasts for 48 h in the presence of 4 μg/mL polybrene.
Determination of ALP activity
ALP activity was determined as described previously (42, 45, 46). In brief, cells were washed with PBS and solubilized with 0.1% Triton X-100, followed by determination of the ALP activity in lysates using p-nitrophenol phosphate as a substrate. Protein contents of the lysates were determined using Bradford protein assay reagent (Bio-Rad, Hercules, CA, USA). For cytochemical analysis, cells were washed with PBS and fixed with 4% paraformaldehyde in PBS. Subsequently, cells were stained with a mixture of 330 μg/mL nitro blue tetrazolium, 175 μg/mL bromochloroindolyl phosphate, 100 mM NaCl, 50 mM MgCl2, and 100 mM Tris (pH 9.5).
Alizarin red staining
Following induction of differentiation, cultured osteoblasts were washed with PBS twice, fixed in 70% ethanol, and stained with 0.4% alizarin red solution for 10 min.
Skeletal preparation of mice
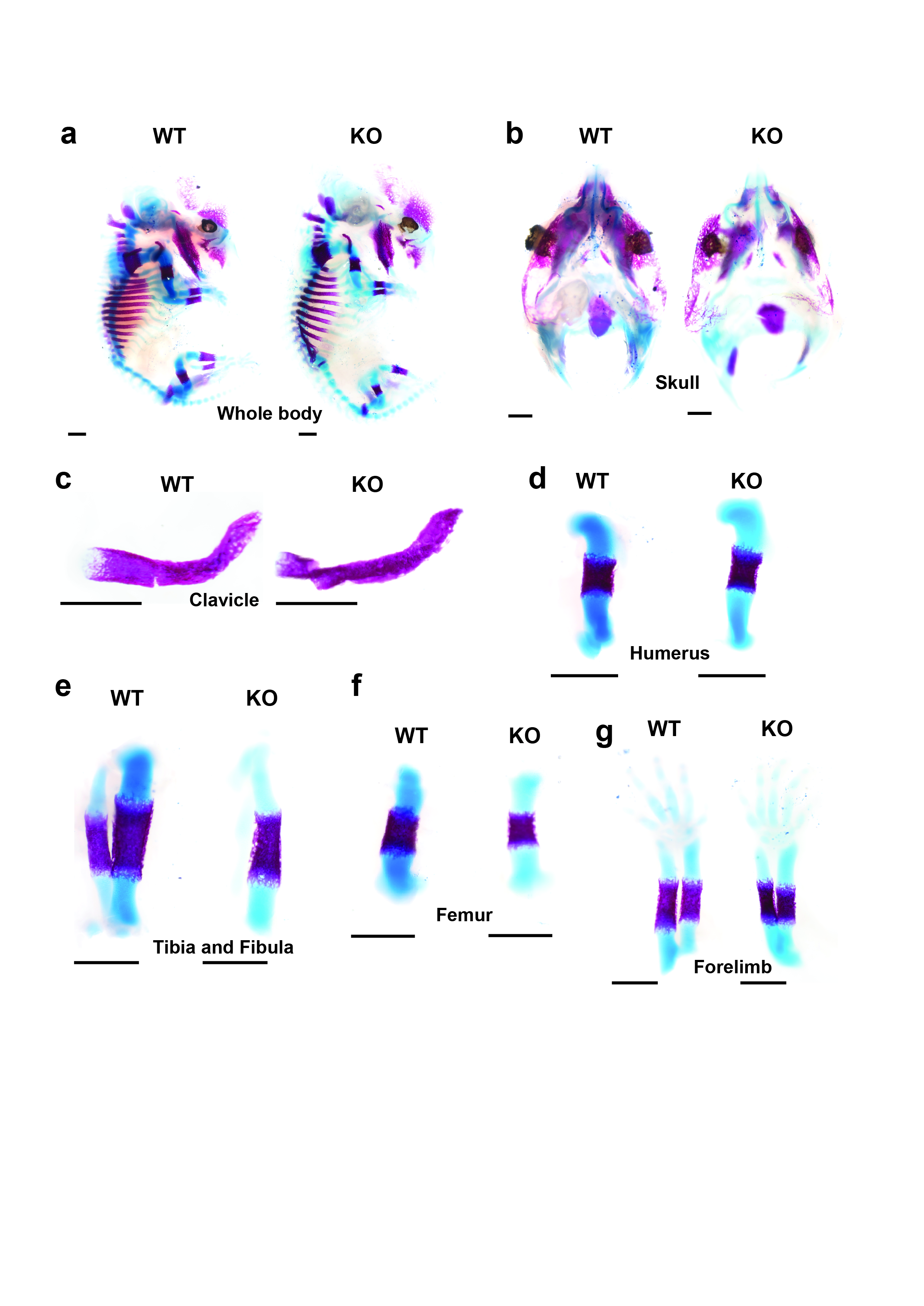
Following removal of the skin and viscera, mice were fixed in 96% ethanol for 24 h. Cartilage was stained for 24 h with alcian blue solution containing 0.015% alcian blue 8GX, 20% acetic acid, and 80% ethanol. Following dehydration with 100% ethanol for 3 days, the whole bodies were digested with 1% KOH at room temperature until the skeleton became clearly visible. The specimens were subsequently stained with 0.002% alizarin red in H2O for 24 h. Finally, the specimens were maintained in 100% glycerol and observed and photographed under an S-APO microscope (Leica, Wetzlar, Germany).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Cultured cells were washed twice with PBS and subjected to total RNA extraction with a Nucleo Spin RNA Plus Kit (Takara). cDNA was synthesized using ReverTra Ace® qPCR RT Master Mix with gDNA Remover (TOYOBO, Osaka, Japan). The individual cDNAs were amplified with THUNDERBIRD® SYBR qPCR Mix (TOYOBO) using a StepOnePlus™ Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The relative expression levels of the target genes was determined by the delta-delta Ct method using transcripts of Actb as the internal reference for each mouse RNA sample. The primer pairs and probes used for amplification were:
Smoc1 Forward, 5′-TGCCTGGGTGTTAGCAAAGAAG-3′, Reverse, 5′-GCGTCCGATGAACGGGTTTG-3′, and Probe, 5′-TGGTAGCCTTGGCAGCTTCCCTCAGG-3′; Smoc2 Forward, 5′-GCTTGGGTGTCACCAGAGAG-3′, Reverse, 5′-CTGGGCTGTCTATTAGAAGAAGAAC-3′, and Probe, 5′-AAGCCAACACCAGGAAGCGCCACA-3′; Osterix Forward, 5′-AGCGACCACTTGAGCAAACAT-3′, Reverse, 5′-GCGGCTGATTGGCTTCTTCT-3′, and Probe, 5′-CCCGACGCTGCGACCCTCC-3′; Osteocalcin Forward, 5′-GCAATAAGGTAGTGAACAGACTCC-3′, Reverse, 5′-GTTTGTAGGCGGTCTTCAAGC-3′, and Probe, 5′-TGGAGCCTCAGTCCCCAGCCCA-3′; Runx2 Forward, 5′-CTCCTTCCAGGATGGTCCCA-3′, Reverse, 5′-CTTCCGTCAGCGTCAACACC-3′, and Probe, 5′-CACCACCTCGAATGGCAGCACGCT-3′; Actb Forward, 5′-TTAATTTCTGAATGGCCCAGGTCT-3′, Reverse, 5′-ATTGGTCTCAAGTCAGTGTACAGG-3′, and Probe, 5′-CCTGGCTGCCTCAACACCTCAACCC-3′.
Whole-mount in situ hybridization
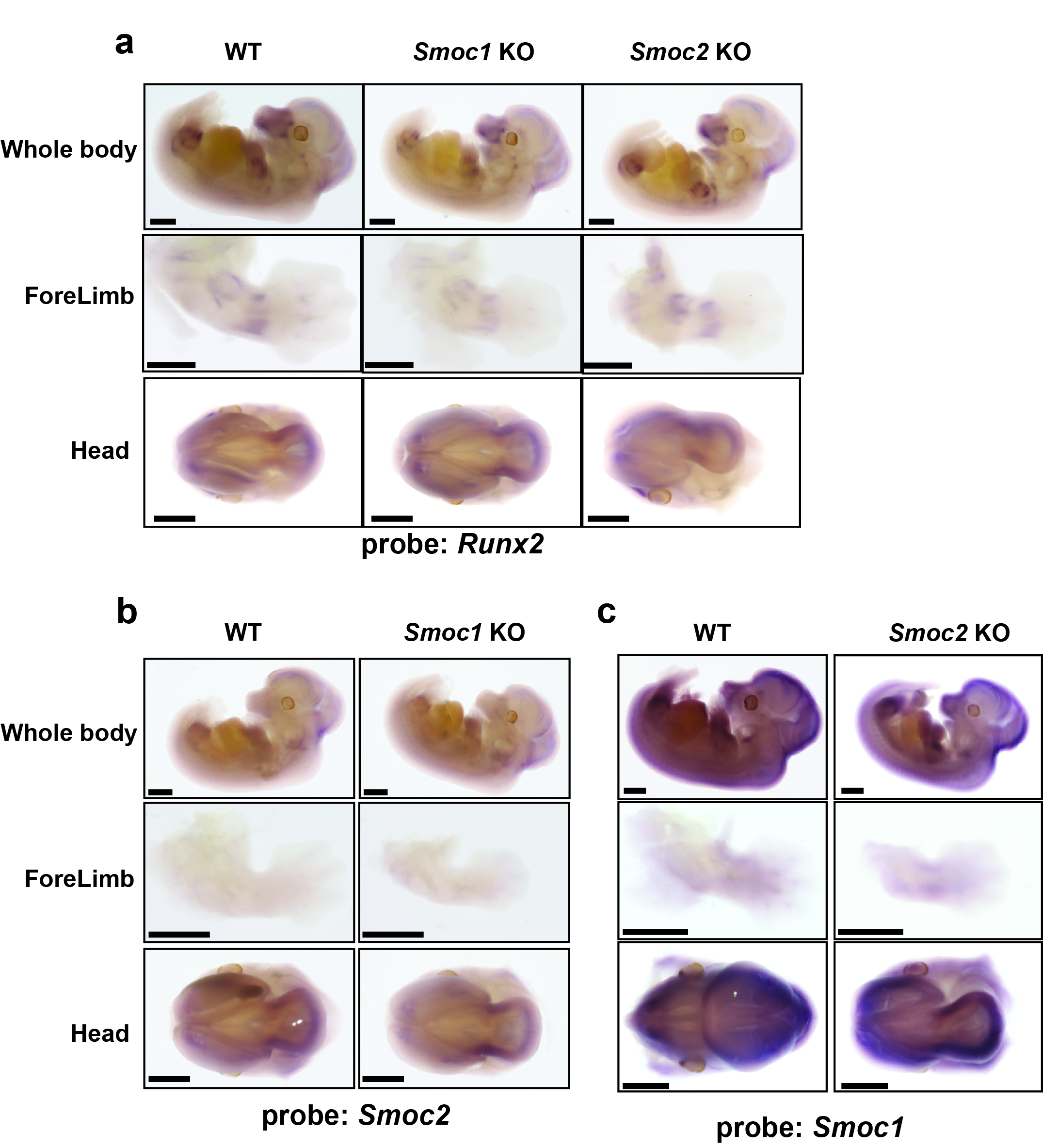
Digoxigenin (DIG)-labeled single-stranded RNA probes were prepared using a DIG RNA Labeling Kit (Roche, Basel, Switzerland). The Smoc1 probe was an 847-bp fragment of the coding sequence (position 386–1232 in the NM_001146217.1 cDNA sequence). The Smoc2 probe was a 1041-bp fragment of the coding sequence (position 460–1500 in the NM_022315.2 cDNA sequence). The Runx2 probe was a 639-bp fragment of the coding sequence (position 3173–3807 in the NM_001146038.2 cDNA sequence).
C57BL6/J mouse embryos (E12.5) were fixed with 4% paraformaldehyde in PBS containing 0.1% Tween-20 overnight at 4 °C. Samples were hybridized with gene-specific DIG-labeled RNA probes overnight at 70 °C, washed, and incubated with 1:2500 diluted anti-DIG-AP Fab fragments (Roche) for 3 h at room temperture. For optimum signal detection, samples were treated with nitroblue tetrazolium chloride/5-bromo-4-chloro-3-indolyl phosphate for various times.
Hematoxilin and eosin (HE) and von Kossa staining
Tissue preparation was conducted as previously described (47). Briefly, mice tibiae at E15.5 were fixed in 4% paraformaldehyde in PBS and used to prepare paraffin-embedded sections with 7-µm thickness. The sections were deparaffinized and stained with Mayer’s hematoxylin and eosin solution. Bone mineralization was analyzed using the von Kossa staining method. In brief, the sections were fixed in 4% paraformaldehyde in PBS and subsequently incubated with 5% silver nitrate for 60 min under UV light. Following rinsing with distilled water, bone nodules were photographed using a phase-contrast microscope.
In situ hybridization
Mice tibiae at E15.5 were fixed in 4% paraformaldehyde in PBS and used to prepare paraffin-embedded sections with 7-μm thickness. DIG-labeled single-stranded RNA probes were prepared using a DIG RNA Labeling Kit (Roche). The Col2a1 probe was a 435-bp fragment of the coding sequence (position 1566–2000 in the NM_031163 cDNA sequence). The Col10a1 probe was a 435-bp fragment of the coding sequence (position 903–1419 in the NM_009925.4 cDNA sequence). The Col1a1 probe was a 324-bp fragment of the coding sequence (position 4466–4789 in the NM_007742.4 cDNA sequence). The Mmp13 probe was a 639-bp fragment of the coding sequence (position 1576–2214 in the NM_008607.2 cDNA sequence). The Pth1r probe was a 780-bp fragment of the coding sequence (position 766–1545 in the XM_017313215.1 cDNA sequence). The Osterix probe was a 1312-bp fragment of the coding sequence (position 36–1347 in the NM_130458.3 cDNA sequence). The sections were mounted on glass slides, successively treated with 10 µg/mL proteinase K and 0.2 M HCl, and subjected to acetylation in 0.1 M triethanolamine/0.25% acetic anhydride. Following prehybridization, the sections were incubated with DIG-labeled RNA probes at 65 °C for 16 h. The sections were further incubated with anti-DIG-AP Fab fragments for 2 h at room temperture. Following washing, the sections were treated with nitroblue tetrazolium chloride/5-bromo-4-chloro-3-indolyl phosphate for various times for optimum staining.
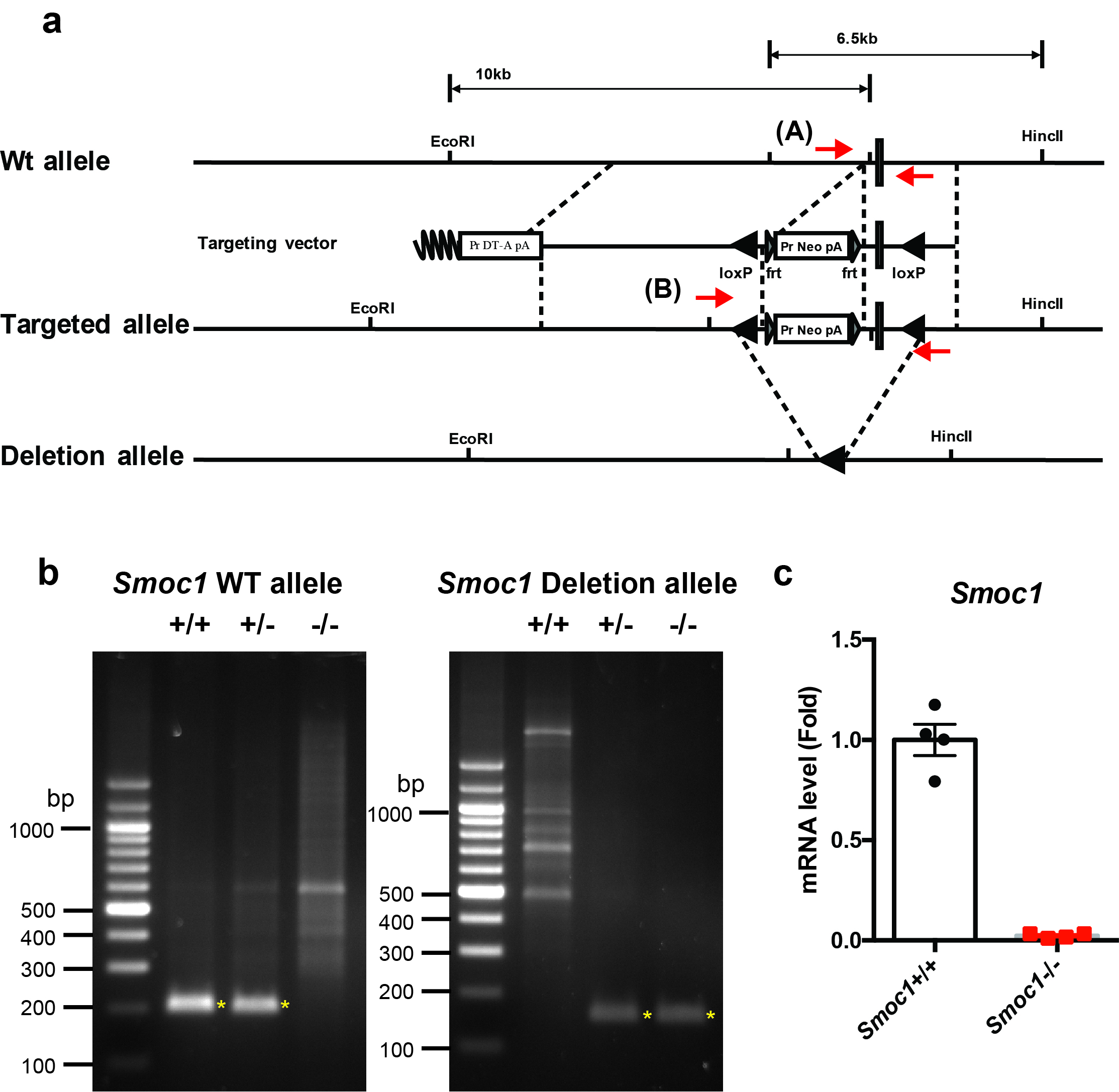
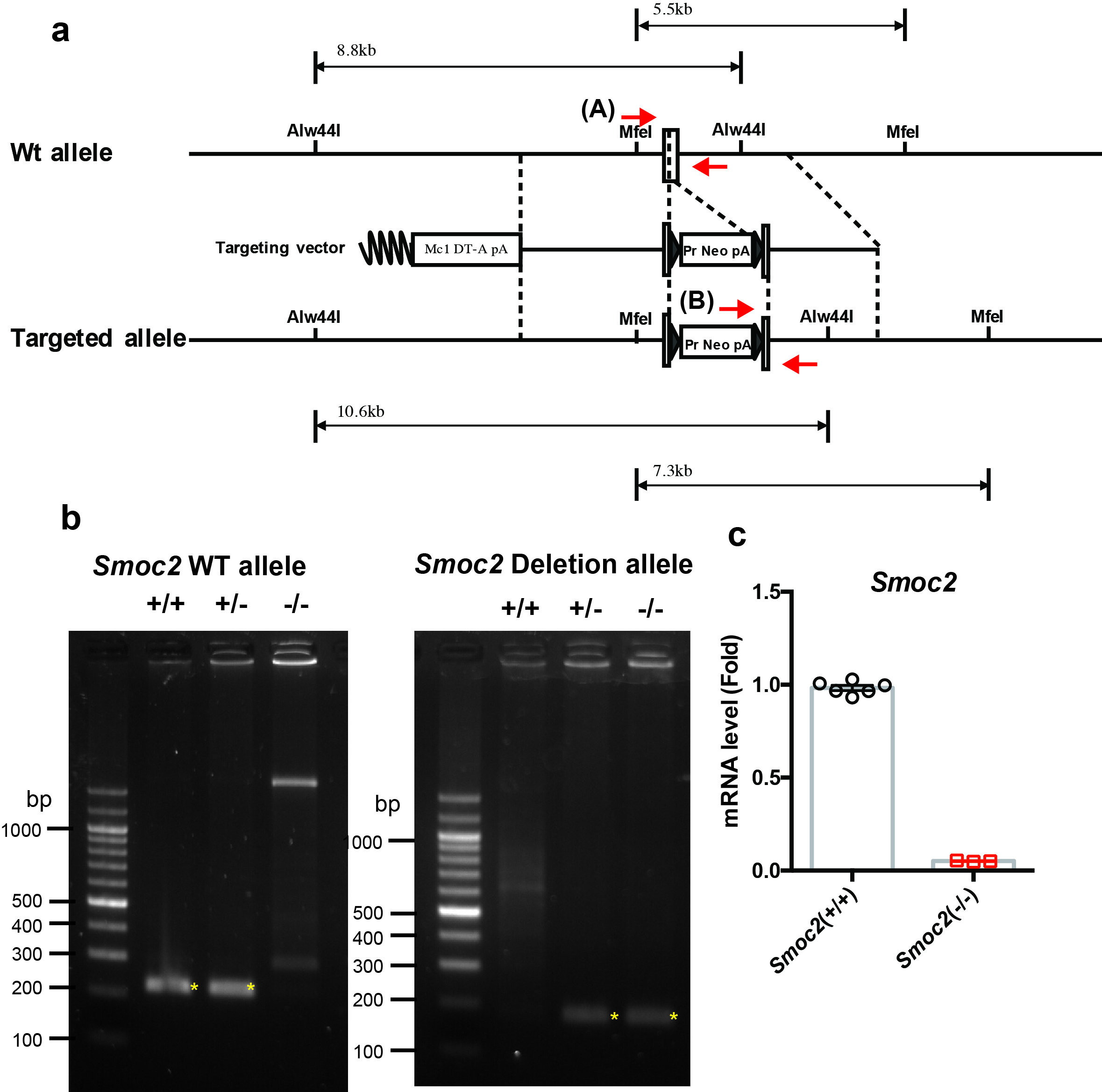
Generation of Smoc1 flox mice and Smoc2 KO mice
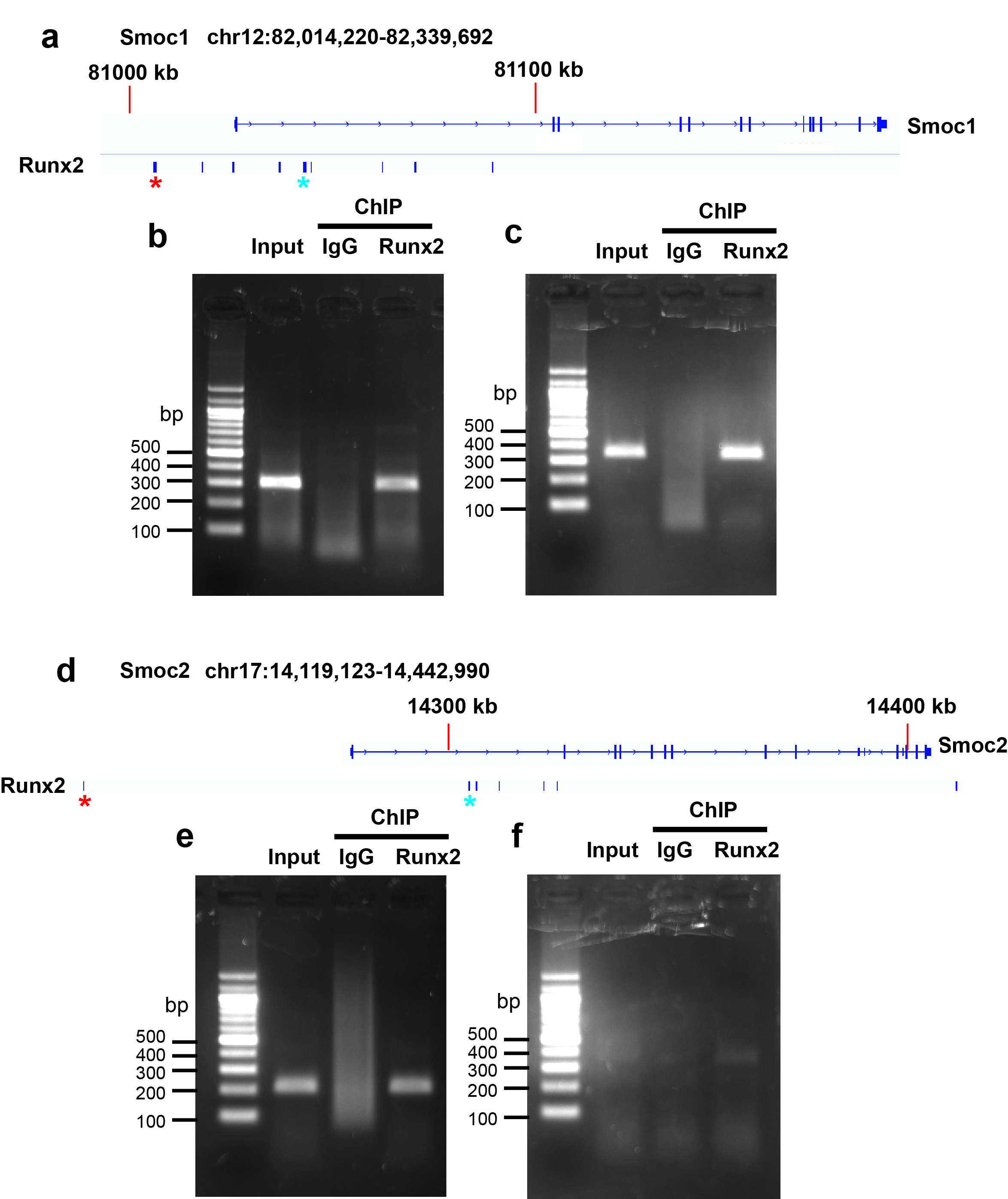
Three splice variants are known to exist for Smoc1 at the protein level according to the Ensemble database (http://www.ensembl.org/index.html). All splice variants contain exon 1 (chromosome 12: 81026808–81027158), exon 2 (chromosome 12: 81104607–81104772), exon 3 (chromosome 12: 81105899–81106011), exon 4 (chromosome 12: 81135776–81135875), exon 5 (chromosome 12: 81137965–81138012), exon 6 (chromosome 12: 81150619–81150708), exon 7 (chromosome 12: 81152673–81152753), exon 8 (chromosome 12: 81167507–81167699), exon 9 (chromosome 12: 81168229–81168311), exon 10 (chromosome 12: 81170186–81170291), and exon 11 (chromosome 12: 81179479–81179723). Exon 1 includes the ATG start codon of Smoc1. Therefore, we decided to generate mutant mice, in which exon 1 of the Smoc1 locus was floxed (Supplementary Fig. 6a). Similarly, three splice variants are known to exist for Smoc2 at the protein level. All splice variants contain exon 1 (chromosome 17: 14279506–14279799), exon 2 (chromosome 17: 14325535–14325706), and exon 3 (chromosome 17: 14336547–14336653). Exon 1 includes the ATG start codon of Smoc2. Therefore, we decided to generate mutant mice, in which exon 1 of the Smoc2 locus was flanked by the PGK-Neo cassette (Supplementary Fig. 7a). These targeting vectors were electroporated into TT2 embryonic stem cells (48) and their homologous recombination was examined by Southern blotting analysis. Germline transmission of the mutant allele of Smoc1 flox or Smoc2 mutant was achieved by mating with C57BL/6J mice and confirmed by Southern blotting and genomic PCR. The accession numbers for the Smoc1 floxed and Smoc2 heterozygous deficient mice are CDB0719K and CDB0802K, respectively (http://www2.clst.riken.jp/arg/mutant%20mice%20list.html). CAG-Cre transgenic mice were provided by the RIKEN Bioresource Research Center (Tsukuba, Japan; RBRC01828). For genotyping, the product sizes were: Smoc1 WT) allele, 202 bp (Forward primer, 5′-TCTCTCCCATTGGCTTCCAC-3′; Reverse primer, 5′-GAGTGCGAGCGTGTGCTCT-3′); Smoc1 deletion allele, 112 bp (Forward primer, 5′-AACCGCCCCTCTCATCTCT-3′; Reverse primer, 5′-GGTCCAGCGACACAACTTTAT-3′); Smoc2 WT allele, 216 bp (Forward primer, 5′-GTTCGCACACCGGATCTTC-3′; Reverse primer, 5′-GGTTCTCAGTGAGGGACAACAG-3′); Smoc2 KO allele, 205 bp (Forward primer, 5′-GTTCGCACACCGGATCTTC-3′; Reverse primer, 5′-GGTTCTCAGTGAGGGACAACAG-3′).
All protocols for the animal use and experiments were approved by the Osaka University Institute Animal Experiment Committee and the Institutional Animal Care and Use Committee of the RIKEN Kobe Branch.
Statistical analysis
The statistical significances of differences in data were determined by two-tailed and unpaired Student’s t-tests for two groups. Difference between three or more groups were compared by one-way analysis of variance (ANOVA) or two-way ANOVA followed by Tukey’s multiple-comparisons test. Values of P < 0.05 were considered to indicate statistical significance.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}