Developing efficient and cost-effective methodologies for high value-added conversion of waste plastic delivers substantial environmental and economic benefits. Herein, we develop a novel approach utilizing boric acid in the methanolysis of waste polyethylene terephthalate (PET) to derive pure dimethyl terephthalate (DMT) and boronic acid esters through in-situ capture of ethylene glycol (EG). It not only upcycles waste PET but also eliminates intricate EG purification processes. Catalyzed by magnalium-aluminum-layered double oxides (Mg4Al1-LDO), this method achieved 100% conversions of PET with 96% and 100% yields of arylboronic esters and DMT, respectively. Kinetic studies and in-situ Fourier-transform infrared spectroscopy (FT-IR) demonstrated the pivotal role of the monodentate methoxy species, generated through the interaction of medium basic Mg–O ion pairs and methanol. This method demonstrates applicability for the upcycling of assorted discarded PET wastes, polyesters, and polycarbonates with EG units, highlighting its potential as a comprehensive solution for waste plastic management.
Article
Enhanced Methanolysis of Waste PET for Sustainable Production of Dimethyl Terephthalate and Cyclic Arylboronic Esters
https://doi.org/10.21203/rs.3.rs-3558735/v1
This work is licensed under a CC BY 4.0 License
Version 1
posted
You are reading this latest preprint version
PET methanolysis
Dimethyl terephthalate
Arylboronic esters
Layered double oxides
Waste plastics
Despite the growing awareness of the environmental crisis caused by excessive plastic consumption and inadequate waste management, plastic production continues to rise in tandem with global economic growth1. It is predicted that by 2050, the environmental accumulation of plastics could exceed 2.5 billion tons2. Polyethylene terephthalate (PET) is one of the most widely used polyester plastics, boasting an annual production of about 70 million tons2,3. Unfortunately, only a small fraction of PET waste undergoes recycling, exacerbating the problem as these waste polyester plastics can break down into microplastic pollutants, causing significant ecological damage4,5. Therefore, developing efficient technologies to convert waste PET carbon resources into value-added commodities has significant environmental and economic benefits. Current PET disposal methods include thermal, mechanical, and chemical recycling6–8. Chemical recycling allows for the transformation of polymer to original monomer, fuel, or other high-value products, thereby amplifying the value chain and product quality6,9.
PET can be chemically depolymerized by nucleophilic agents to cleave the ester group towards monomers or their derivatives10–14. Hydrolysis and alcoholysis are the most widely used methods to recycle monomers for new PET syntheses15–17, and extensive studies are carrying out to improve the efficiency and reduce the cost18–23. Parallel to this closed-loop recycling, a more promising strategy is to take use of the structural characteristics of waste PET as feedstock for more valuable chemicals. High-selectively transforming the original building blocks is the key for the effective construction of target high-value products. For example, by catalytically hydrogenating ester group directly or after solvolysis, a range of value-added chemicals could be produced, including alcohols15,24, aromatics10,16,25, and carboxylic acid derivatives12. However, keeping TPA or DMT units is still highly desired due to their great market demand as PET monomers. In addition, the high boiling point and strong hydrogen-bonding capacity of ethylene glycol (EG) escalates the energy consumption of purification, especially in aqueous systems. This necessitates exploring methods to upgrade EG into more valuable chemicals, thereby preserving high-value TPA/DMT while bypassing the arduous purification of EG. The most widely reported approaches are partial oxidation of EG into formic acid or its salts17,26–29. However, the product selectivity in these coupled reductions and oxidations process is sensitive to the catalysts, and the value of the product is not significantly improved compared with EG. More valuable and facile routes to upgrade waste PET based on its building units are desperately to be developed.
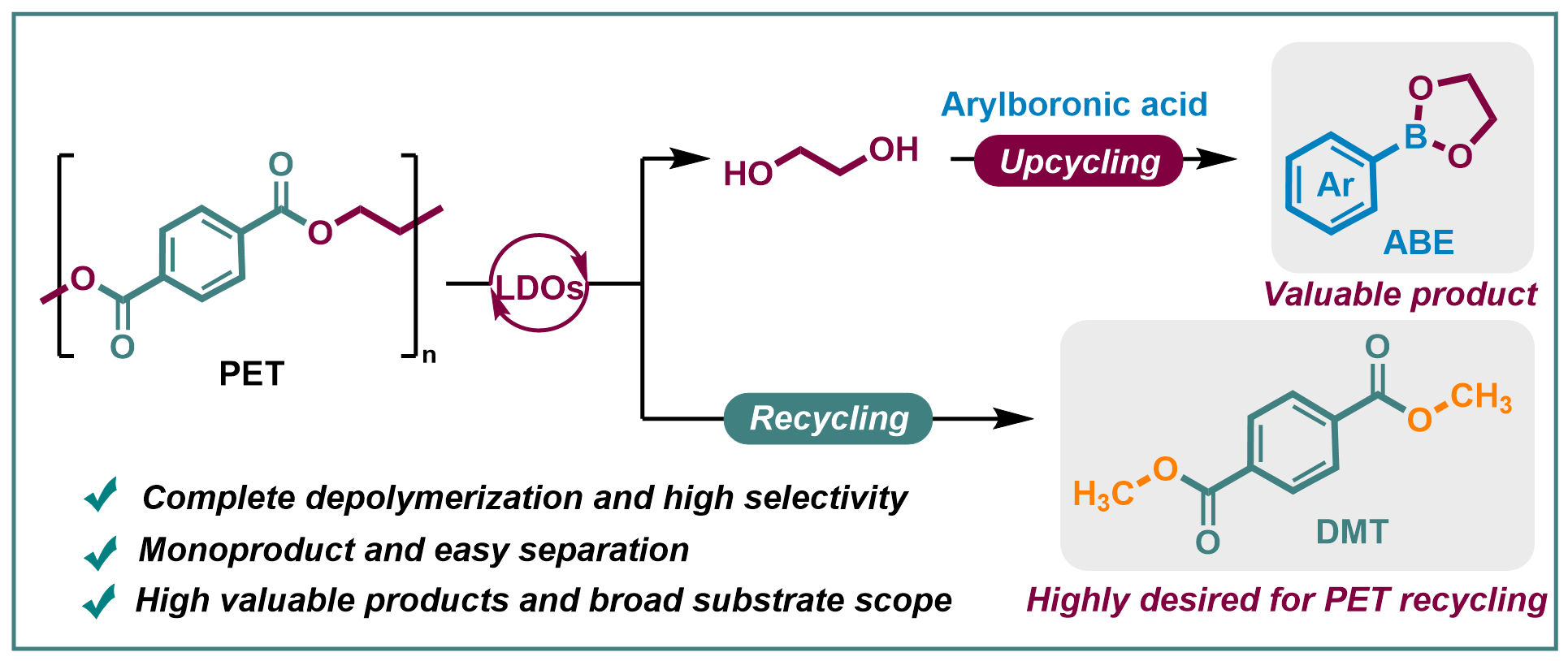
Herein, we pioneer a facile EG upcycling strategy coupled with PET methanolysis, enabling the synthesis of diverse five-membered arylboronic esters (ABE) and DMT (Scheme 1). Arylboronic esters are important organic boron compounds widely used in the efficient construction of pharmaceuticals and natural products30–34, which are typically obtained by reversible binding of diols and boric acid. By simply introducing arylboric acid into the methanolysis system of waste PET, the reaction can proceed completely and the utilization value of EG is significantly improved by upgrading to valuable arylboronic esters. The process, facilitated by basic layered double oxides (LDOs), successfully achieved 100% conversions of PET and boric acid with 96% and 100% production yields of arylboronic esters and DMT, respectively. This facile strategy holds great potential for PET upcycling towards DMT and five-membered ring boronic acid esters. It avoids the issue of uncontrollable selectivity that arises in previous oxidation/reduction reactions. The precipitated DMT crystals can be easily separated and purified by recrystallization to be used as high-quality monomer for new PET; the dissolved boronic acid esters can be easily purified through evaporative crystallization for further use in organic synthesis. Overall, this work paves a new way for effective recycling of waste polyester plastics, owing to its advantages such as complete utilization and high selectivity, easy separation and purification, and highly valuable products with broad substrate scope.
3.1 Catalytic System Exploration
The devised methodology for waste PET upcycling involves PET methanolysis into DMT + EG and EG capture via esterification with arylboric acid to afford ABE. The initial experiment conducted without a catalyst resulted in limited depolymerization of PET waste, with only 8% DMT and 10% ABE yield after operating at 180°C for two hours. Meanwhile, the toluene byproduct was generated undesirably with the yield of 19%, which was attributed to the rapid protodeboronation of arylboric acid (Ar-B → Ar-H) with the assistance of water35,36. As acid or base can catalyze both methanolysis and esterification reaction, some typical acids and bases were tested and yields of DMT and ABE heavily depended on the catalyst used. Figure 1a demonstrated that acidic Amberlyst-15 exhibited negligible promotion towards PET methanolysis, resulting in almost unchanged DMT and ABE yields. Conversely, basic Mg(OH)2 and magnesium-aluminum layered double hydroxides (Mg-Al-LDHs) enhanced the production of DMT and ABE. Notably, under identical conditions (180°C, 2 hours), Mg4-Al1-LDH achieved remarkable yields of 60% for ABE, 58% for DMT, along with a toluene byproduct yield of 28%. However, incomplete PET depolymerization and undesired protodeboronation of p-tolylboronic acid into toluene were still observed. This issue is likely due to the inadequate basicity of Mg-Al-LDH and the presence of copious interlayer water molecules and hydroxide ions. Previous studies have confirmed that the expulsion of interlayer water molecules and hydroxide ions from LDHs by high-temperature calcination results in the formation of layered double oxides (LDOs) with stronger basicity37,38. Thus, we synthesized Mg4-Al1-LDOs with various calcination temperatures to mitigate the effects of water and hydroxide ions in hydrotalcite layers. As anticipated, calcination significantly improved ABE and DMT yields, and increasing calcination temperatures proved favorable for PET depolymerization (Fig. 1b). When the calcination temperature was 500°C, the yield of ABE and DMT reached 96% and 100%, respectively, effectively suppressing the protodeboronation of p-tolylboronic acid. The influence of the Mg/Al molar ratio in Mg-Al-LDOs was also investigated (Fig. 1c). A lower Mg/Al ratio resulted in weakened PET depolymerization and reduced yields of ABE and DMT. LDOs containing different divalent metals (Zn2+, Ni2+, and Cu2+) were tested as well, but these alternatives resulted in lower ABE and DMT yields (Fig. 1d), due to their weaker basicity. Furthermore, we examined the effect of catalyst loading. Incomplete PET depolymerization occurred when insufficient amount of catalyst was used (Fig. 1e). Increasing catalyst loading proved beneficial for both PET methanolysis and arylboric acid esterification. With a catalyst loading of 0.03 g, both PET and p-tolylboronic acid were entirely converted into ABE and DMT, yielding 96% and 100%, respectively, at 180°C for 2 hours (Fig. 1e). Adjusting the reaction temperature resulted in lower ABE yields (Fig. 1f). Operating the Mg4-Al1-LDO at a lower temperature of 140°C only yielded 33% ABE and 29% DMT for 2 hours (Fig. 1f). In contrast, an elevated temperature of 200°C promoted protodeboronation of p-tolylboronic acid to produce toluene side product (Fig. 1f). Moreover, when scaling up the reaction five times under optimal condition, we also achieved high yields of the five-membered cyclic product ABE (93%, 3a in Fig. 2).
Subsequently, we tested the compatibility of the system with various arylboronic acids. Encouragingly, the system accommodated arylboronic acids bearing different functional groups on the benzene ring, affording various five-membered annulation arylboronic esters 3b-3f in 80–97% yield and DMT in 97–99% yield (Fig. 2). The system demonstrated excellent tolerance of the electronic properties of arylboronic acids, regardless containing electron-donating (-OMe) or electron-withdrawing groups (-F, -CO2Me), or the position of the substituent on the benzene ring (-m-Me). Furthermore, 1-naphthylboric acid also displayed high reactivity and successfully reacted with PET to provide the cyclic product 3g in a satisfactory yield (80%). Intriguingly, other waste plastics with adjacent diol units, such as poly(ethylene succinate) (PES) and poly(ethylene adipate) (PEA), and polycarbonates like poly(propylene carbonate) (PPC), which are widely used across various fields, were viable candidates for our process. They can be converted into corresponding borate esters and diesters with impressive yields (Fig. 2, Nos. 1–3) under standard conditions. To further validate the practicality of this new method, we tested varieties of waste commercial PET samples, including woven tape, woven mesh, transparent film, and non-woven fabric. Excitingly, ABE and DMT were harvested with high yields (Fig. 2, Nos. 4–6, 8). Moreover, this catalytic system can also be effectively applied in the upcycling of twelve different waste PET bottles. All tests demonstrated high yields of both ABE and DMT, regardless previous use or disposal status of the bottles (Fig. 2, No. 7 and Supplementary section S3 for detailed data).
3.2 Characterization of catalysts.
X-ray diffraction (XRD) experiments were performed to confirm the structure of the catalysts and the patterns are shown in Fig. 3a. Strong (003), (006), (009), (110), (113) and broadened (015), (018) reflections were observed for the Mg4-Al1-LDH sample (Fig. 3a), which are the characteristic diffraction peaks of hydrotalcite-like compounds39. Upon calcination at high temperatures (400, 500, and 600°C), the hydrotalcite-like solids undergo decarbonation and dehydration processes, forming mixed metal oxides (Mg4-Al1-LDO). All the calcinated Mg4-Al1-LDOs exhibit the peaks similar with periclase MgO, characterized by the peaks at 43° and 63° with no residual traces of the hydrotalcite phase37,39. No phases containing aluminum were detected, indicating that Al3+ is highly dispersed in a mixed oxide phase. This result shows that the hydrotalcite-like structure of Mg4-Al1-LDH has been fully converted into the mixed oxide phase of Mg4-Al1-LDO by destroying the layered structure under high temperature (400–600°C). In order to obtain a more in-depth understanding of the structural changes that occur during calcination, 27Al MAS NMR analysis was performed. Figure 3b shows the 27Al MAS NMR spectra of Mg4-Al1-LDH and Mg4-Al1-LDO calcined at different temperatures ranging from 400 to 600°C. As anticipated, the spectrum of fresh Mg4-Al1-LDH exhibited a single resonance near 9.9 ppm, which can be assigned to octahedrally coordinated Al40. This data confirm that the LDH structure is formed by metals coordinated to six hydroxyl groups with octahedral geometry. However, when the samples were calcined at 400°C, a new signal at around 80 ppm is observed. Meanwhile, the six-coordinated Al resonance is quite broad and shifts slightly to 13 ppm. These can be attributed to the formation of tetrahedrally coordinated Al. This suggests that after calcination, the Al coordination partially changes from six-coordinated to four-coordinated due to the removal of CO32- and H2O. Further increasing calcination temperature to 500 or 600°C, both the four- and the six-coordinated Al peaks broaden further and shift slightly to 76 ppm and 15 ppm. These results indicate that the calcination process leads to the transformation from the hydrotalcite structure to mixed oxide phase.
As the catalytic activity heavily depends on the basicity of the catalysts, we employed temperature-programmed desorption of CO2 (CO2-TPD) to characterize the basicity property. Based on previous literature, we can categorize CO2 desorption temperatures (Td) below 200°C as weak Brönsted basic sites, which correspond to surface hydroxyls on the metal oxide41. Medium Lewis basic sites (300–450°C) are associated with metal-oxygen pairs, such as Mg–O ion pairs. Strong Lewis basic sites (500–650°C) are related to CO2 desorption on the surface oxygen anions of low coordination37,42. The uncalcined Mg4-Al1-LDH exhibits a significant peak at 420°C (Figure S3a), attributing to the existence of terminal Brönsted OH– basic sites43. They are responsible for the undesired protodeboronation of p-tolylboronic acid35,36, yielding 28% toluene (Fig. 1b). After calcination, the Brönsted OH– basic sites decrease due to the removal of surface hydroxyl groups, meanwhile alternative basic sites form. The variation of the basic sites can lead to different catalytic pathways, which may exhibit distinct selectivity and reactivity. Therefore, it is crucial to carefully consider the impact of calcination on the catalytic properties. All Mg4-Al1-LDOs with varying calcination temperatures predominantly exhibit weak and medium basic sites, as indicated in (Fig. 3d). With a calcination temperature at 400°C, Mg4-Al1-LDO400°C produces ABE and DMT with 77% and 100% yield, respectively, along with a 23% yield of toluene, outperforming Mg4-Al1-LDH due to the eradication of Brönsted OH– basic sites (Fig. 1b). Raising the calcination temperature from 400°C to 600°C, the CO2 adsorption peaks shift to lower temperatures and become broader, transitioning from a sharp single peak to broad double peaks. It suggests a decrease in the strong base capacity and an increase in weak and medium base capacity (Fig. 3d). As a result, the reaction selectivity using Mg4-Al1-LDO500°C and Mg4-Al1-LDO600°C were improved, with 4% and 11% yeild of protodeboronation product, respectively (Fig. 1b). To further understand the influence of basic strength on the catalytic activity, we investigated LDOs with different metals. Figure 3e shows that the Ni4-Al1-LDO, Zn4-Al1-LDO, and Cu4-Al1-LDO catalysts have significantly less basicity compared to the Mg-Al-LDOs. These LDOs mainly contain weak basic sites with negligible medium and strong basic sites. They exhibited underwhelming ABE and DMT yields, even comparable to the catalyst-free condition. We posit that weak basic sites may lack the ability to catalyze PET depolymerization effectively. This suggests that medium basic Mg–O ion pairs are the actual active sites. To verify this, Mg-Al-LDO with different Mg/Al molar ratio were studied (Figure S1a). As the Mg/Al ratio increases from 2 to 4, the peak area of medium basic sites versus weak basic sites also increase. Mg2-Al1-LDO mainly has weak basic sites and yield 81% ABE and 76% DMT (Fig. 1c). Mg3-Al1-LDO has considerable medium basic sites but is still dominated by weak basic sites, resulting in increased ABE and DMT yields of 94% and 91%, respectively (Fig. 1c). Mg4-Al1-LDO possesses more medium basic sites, leading to 96% and 100% yields of ABE and DMT (Fig. 1c) respectively, despite having a similar total basicity content to Mg3-Al1-LDO. These results highlight the critical role of medium basic Mg–O ion pairs in the LDOs for both the reaction activity and selectivity.
3.3 Reaction Kinetics
To comprehend the reaction pathway, we examined the reaction kinetics. The results show that the yields of ABE and DMT in the overall reaction increase continuously with the reaction progress, reaching their maximum levels of 96% and 100%, respectively, by the 2-hour mark (Fig. 4). As previously discussed, the production of ABE through PET depolymerization involves the methanolysis of PET followed by the esterification of EG and boric acid. However, we did not detect any EG monomer throughout the entire reaction process. It has been reported that the esterification of EG and boric acid occurs rapidly30,31, which implies that any produced EG is instantly esterified, thereby accounting for its absence during the reaction process. To confirm this, we investigate the tandem reactions separately under identical reaction conditions, i.e., the methanolysis of PET (Reaction 1) and the esterification of EG with boric acid (Reaction 2). The yields of DMT and ABE versus reaction time reflect the reaction rates of methanolysis and esterification. As shown in Fig. 4, under catalysis of Mg4-Al1-LDO, boric acid and EG (Reaction 2) rapidly dehydrate and cyclize, leading to an 80% yield of ABE within 10 minutes, and a complete conversion of the feedstock within 20 minutes. In contrast, the methanolysis of PET, in the absence of arylboric acid, results in a modest DMT yield of 8% after 20 minutes. It shows that the esterification of boric acid with EG proceeds (Reaction 1) significantly faster than the methanolysis of PET (Reaction 2). It is noteworthy that the reaction kinetics curve for PET methanolysis and the overall reaction are nearly identical, suggesting that the methanolysis of PET is the rate-determining step of this system. Intriguingly, in the initial 40 minutes of the reaction, DMT produces in overall reaction faster than sole PET methanolysis in reaction 1 under the same reaction condition. It indicates that the cyclization of boric acid with EG notably accelerated the PET methanolysis to produce DMT.
3.4 Reaction Mechanism.
According to the findings discussed above, medium basic Mg–O ion pairs play a crucial role in the reaction pathways of PET depolymerization. To shed light on the rate-determining methanolysis step and get insight into the reaction mechanism, we conducted in situ IR spectroscopy analysis. The depicted spectra signify the difference IR spectra with the pre-heated sample serving as the reference point. Methanol adsorption on Mg-Al-LDO at various temperatures (Fig. 5b) with different Mg/Al ratios (Fig. 5c) was studied. The interaction between methanol and the catalyst surfaces induces changes in two primary regions, 3100 ~ 2700 cm− 1 and 1200 ~ 1000 cm− 1, which corresponds to the ν(CH3) and ν(O-C) vibration mode of methanol and/or methoxy species, respectively. Post-methanol adsorption IR spectra of LDOs reveal the formation of three types of active adsorbed species, each with distinct (O-C) vibrations. These include non-dissociated methanol (H species, ~ 1030 cm− 1), monodentate methoxy species (Type I, ~ 1100 cm− 1), and bidentate methoxy species (Type II, ~ 1060 cm− 1) 39,44, as shown in Fig. 5a.
Upon introduction of methanol via argon bubbling, three types of intermediates emerge, with bidentate species Type II at 1060 cm− 1 and non-dissociated methanol at 1030 cm− 1 dominating, and a weak band for monodentate species Type I at 1100 cm− 1. As the methanol adsorption time increases, the band area of all intermediate species increases, suggesting an enhanced interaction between the Mg–O active site and methanol (Supplementary Figure S2). The band areas of the adsorbed methanol stopped increasing after 15 minutes, indicating saturation of methanol adsorption. We then compared the infrared spectra of Mg4-Al1-LDO with adsorbed methanol at saturation (20 min) at different temperatures (140°C, 180°C, and 200°C). As shown in Fig. 5b, the band area of Type II and H species intermediates were comparable, while Type I displays a distinctly different band area that increases with temperature. Notably, the largest band area of Type I occurs at 180°C, which aligns with the PET depolymerization activity shown in Fig. 1f. The effect of the Mg/Al ratio has a similar pattern across all three catalysts. While the band areas of Type II and H species were comparable, Type I shows significant differences (as shown in Fig. 5c), increasing with the Mg/Al ratio and correlating with the order of PET depolymerization activity (Fig. 1c). This indicates that Type I plays a crucial role in the reaction, as it has been reported to be the active intermediate in the methanol transesterification reaction39. To further verify the relationship between Type I adsorption mode and reaction activity, Table 1 outlines the deconvoluted band area for LDOs with different Mg/Al ratios and temperatures, normalized by catalyst weight. The results show that DMT yield is positively associated with methoxy Type I area, suggesting a direct link between Type I methoxy species and PET methanolysis. To further explore the function of Type I monodentate species in the PET methanolysis process, we analyzed the reaction with in-situ IR spectroscopy by introducing PET as a reactant. Mg-Al-LDO with different Mg/Al ratios were mixed with PET in a 3:1 mass ratio. Notably, for all catalysts at 180°C, the presence of PET results in complete disappearance of the band at 1100 cm− 1 after methanol adsorption, while the other methoxy bands remained almost unaltered (as shown in Fig. 5d-f). This observation confirms that methoxy Type I can react quickly with PET, underscoring its pivotal role in the reaction.
Building upon reported work45 and our current experimental findings, we propose a plausible mechanism for this chemical upcycling process, as depicted in Fig. 6. Initially, Mg–O in Mg-Al-LDO activates methanol by extracting its protons, forming CH3O–. This CH3O– intermediate then functions as a nucleophilic reagent, attacking the C = O of PET and leading to a tetrahedral intermediate, which gradually breaks down the long chain to generate DMT and EG, facilitated by H+ on the surface of the LDOs. As the molecular chain shortens and molecular weight reduces, DMT, PET oligomers, and EG fragments are generated due to efficient cleavage of the ester C = O bond. The oligomers then rapidly decompose, leading to an increased yield of DMT and EG fragments. Concurrently, the in-situ produced EG is swiftly captured by arylboric acid, aided by Mg4-Al1-LDO, during the methanolysis process. This sequence leads to a dehydration cyclic reaction, yielding ABE product.
3.5 Stability of Mg4-Al1-LDO for PET upcycling
The longevity and robustness of heterogeneous catalysts are crucial for their industrial application. To evaluate the stability of the Mg4-Al1-LDO catalyst, multiple experimental cycles involving a dichloromethane wash were conducted. The catalyst demonstrates consistent high levels of activity in PET methanolysis coupled with the dehydration cyclization of boric acid for up to four cycles, showing negligible degradation (Fig. 7a). A steady decline in ABE yield was observed after four cycles, yet the DMT yield appeared to be minimally impacted. These results indicate that PET methanolysis remained unaffected, while the protodeboronation of p-tolylboronic acid was significantly intensified. As previously noted, the deboronation process occurs in a system containing H2O and OH–. Our analysis indicates that the H2O originates from the esterification reaction between EG and boric acid.
To trace the source of OH–, the catalyst underwent a series of characterization methods after being cycled six times (Supplementary Section S4). The XRD analysis revealed that the cycled Mg4-Al1-LDO gradually transits back to the LDHs structure (Fig. 7b). This transition, caused by the absorption of water during the reaction and air-drying processes, suggests that the OH– within the reformed LDHs structure serves as the active site for the protodeboronation of p-tolylboronic acid. This catalytic activity leads to decrease of ABE yield from 96–81%. It appears, therefore, that catalyst inactivation is linked to both the H2O present in the reaction system and the OH– within the catalyst. The gradual reversion to the layered hydrotalcite structure also necessitates H2O, suggesting that the introduction of an inert drying agent to remove H2O could efficiently inhibit the deboronation side reaction. The deactivated catalyst was subjected to high-temperature calcination for catalyst regeneration, effectively restoring the Mg4-Al1-LDO structure. Upon reuse in the reaction cycle, this regenerated catalyst successfully produced ABE and DMT in yields of 94% and 100%, respectively (Fig. 7a), affirming that the catalytic efficacy of the recycled Mg4-Al1-LDO is successfully restored.
In this study, we have proposed an effective and facile method for the upcycling of waste PET to afford various value-added five-membered cyclic arylboronic esters and dimethyl terephthalate (DMT) in optimal yields. Facilitated by the Mg4-Al1-LDO catalyst, the reaction demonstrates a complete conversion of PET and boric acid, yielding 96% and 100% of arylboronic esters and DMT, respectively. Kinetic study reveals that the rate-determining step in this entire process is the methanolysis of PET. In-situ Fourier-transform infrared spectroscopy (FT-IR) demonstrates that methanol adsorbed on the medium basic Mg–O ion pairs, and the resulted monodentate methoxy species plays a pivotal role in the catalytic process. The catalyst can be recycled and easily regenerated. This method can be successfully extended to recycle various types of waste PET, offering a pragmatic solution for high-value chemical recycling of plastic waste. It could potentially mark a significant step in addressing the global plastic waste challenge and pave a new way for more efficient and environmentally friendly recycling methods.
Catalyst preparations
Layered double hydroxides (LDHs) were prepared via a co-precipitation method at a constant pH of 10. Two solutions were prepared as follows: Flask A contained NaNO3 (6.80 g, 0.08 mol) dissolved in 50 mL of DI water, while Beaker B contained Mg(NO3)2·6H2O (15.38 g, 0.060 mol) and Al(NO3)3·9H2O (5.63 g, 0.015 mol) dissolved in 50 mL of DI water. The solution in Beaker B was added dropwise to Flask A, while maintaining a controlled pH of 10 using a 1 M NaOH solution. The reaction was conducted at ambient temperature for 2 hours, followed by thermal treatment of the solution in Flask A. The solution was heated to 80°C with concurrent stirring and refluxing for a total of 12 hours. The resulting precipitate was filtered, washed with hot deionized water, and dried in a vacuum drying oven at 80°C for 24 hours, leading to the formation of double hydroxide (Mg4-Al1-LDH) 37,39. The obtained Mg4-Al1-LDH was ground into a powder and calcinated under an Ar flow in a tube furnace at different temperatures (400°C, 500°C, and 600°C) for 8 hours. The final products were named Mg4-Al1-LDOx°C, where x represents the calcination temperature. If there was no special emphasis, the calcination temperature was 500°C. The synthesis procedures of Mg3-Al1-LDO and Mg2-Al1-LDO were similar to Mg4-Al1-LDO by adjusting the Mg/Al molar ratio. Similarly, the synthesis procedures of Zn4-Al1-LDO, Ni4-Al1-LDO, and Cu4-Al1-LDO were similar to Mg4-Al1-LDO by adjusting the divalent metal of the feedstock46–48.
Activity tests
PET powder (1 mmol), p-tolylboronic acid (1 mmol), and Mg4-Al1-LDO (0.03 g) were added to an autoclave containing 5 mL of MeOH. The autoclave was then stirred at 180°C for 2 hours. After completion of the reaction, the reaction solution was diluted with 10 mL of dichloromethane. The liquid products were analyzed by GC using mesitylene (0.5 mmol) as an internal standard. The depolymerization of other polyesters and polycarbonates, including poly(ethylene succinate) (PES), poly(ethylene adipate) (PEA), and poly(propylene carbonate) (PPC), was carried out in the same manner as the depolymerization of PET.
Characterizations
Powder X-ray diffraction (XRD): The crystal structure of the sample was characterized using a Bruker D8 Advance diffractometer (Cu-Ka radiation 1.540598A). The scanning parameters were: Cu-Kα X-ray source, λ = 1.5406 Å, tube voltage of 40 kV, tube current of 40 mA, scanning range of 5–90°, scanning speed of 5°·min–1, and scanning step of 0.02°.
1 H-NMR and 13C-NMR spectra were recorded at room temperature using a Bruker Avance-600 instruments (1H NMR at 600 MHz and 13C NMR at 151 MHz), NMR spectra of all products were reported in ppm with reference to solvent signals [1H NMR: CD(H)Cl3 (7.26 ppm), 13C NMR: CD(H)Cl3 (77.00 ppm)]. Signal patterns are indicated as s, singlet; d, doublet; dd, doublets of doublet; t, triplet, and m, multiplet. The detailed NMR characterizations and spectra of product arylboronic esters are provided in Supplementary section S5 and S6.
The product yield was analyzed by Gas chromatography (GC): Agilent 8860 with Agilent J&W HP-5 Polysiloxane GC Column and Gas chromatography-mass spectrometry (GC-MS): Agilent 7890A/5975C GC/MSD with Agilent J&W HP-5 Polysiloxane GC Column.
CO2-TPD study was conducted by a TP-5079 chemisorption equipment (Xianquan, China) equipped with a Mass spectrometry (MS) (Hiden Analytical Ltd, UK). Before the detection step, the sample was treated in helium flow (50 mL/min) at 300°C for 1 h. Subsequently, the sample was cooled down to 50°C, and adsorbed with pure CO2 for 0.5 h. Then the sample was flashed in helium flow (50 mL/min) at 50°C for 2 h for removing physical adsorbed CO2. Finally, TPD analysis was performed at a heating rate of 10°C /min from 50°C to 800°C.
In-situ DRIFT spectra were recorded at a spectral resolution of 2 cm− 1 using a Nicolet iS20 FT-IR spectrometer (Thermo Fisher Scientific). The IR spectra of adsorbed methanol were obtained using a quartz IR cell sealed with CaF2 windows connected with a vacuum system. For removing impurities from the catalyst surface, a pre-treatment was carried out under a N2 flow (50 cm3 STP/min) at 450°C for 1 h. Next, the system was cooled down to the specific temperature (140°C, 180°C or 220°C) and the spectra of samples after pretreatment were correspondingly used as background. Then methanol adsorption was carried out at this specific temperature for different times (1, 5, 10, 15, and 20 min) using a stream of N2 saturated with methanol vapor at room temperature and the corresponding spectra were collected.
Acknowledgment
This work was supported by National Natural Science Foundation of China (22209146, 22376183, 22293015) and the Fundamental Research Funds for the Central Universities (226-2023-00041, 226-2023-00077).
Author contributions
Minghao Zhang: experiments perform, formal analysis, and writing-original draft. Yunkai Yu: experiments perform, formal analysis, graphic design, writing. Binghui Yan: formal analysis. Yu Liu: NMR analysis. Weixiang Wu: formal analysis. Xiuju Song: formal analysis and resources. Yixiong Feng: writing. Bailiang Chen: discussion and writing revision. Buxing Han: writing and resources. Qingqing Mei: conceptualization, formal analysis, funding acquisition, resources, supervision and writing.
Competing interests
The authors declare no competing interests.
- MacLeo, M., Arp, H. P. H., Tekman, M. B. & Jahnke, A. The global threat from plastic pollution. Science 373, 61-65 (2021).
- Geyer, R., Jambeck, J. R. & Law, K. L. Production, use, and fate of all plastics ever made. Sci. Adv. 3, 5 (2017).
- Wu, X. Y., Galkin, M. V., Stern, T., Sun, Z. H. & Barta, K. Fully lignocellulose-based PET analogues for the circular economy. Nat. Commun. 13, 12 (2022).
- Seeley, M. E., Song, B., Passie, R. & Hale, R. C. Microplastics affect sedimentary microbial communities and nitrogen cycling. Nat. Commun. 11, 10 (2020).
- Rillig, M. C. & Lehmann, A. Microplastic in terrestrial ecosystems. Science 368, 1430-1431 (2020).
- Martín, A. J., Mondelli, C., Jaydev, S. D. & Pérez-Ramírez, J. Catalytic processing of plastic waste on the rise. Chem 7, 1487-1533 (2021).
- Chu, M. Y., Liu, Y., Lou, X. X., Zhang, Q. & Chen, J. X. Rational design of chemical catalysis for plastic recycling. ACS Catal. 12, 4659-4679 (2022).
- Peng, Y. et al. Acetolysis of waste polyethylene terephthalate for upcycling and life-cycle assessment study. Nat. Commun. 14, 3249 (2023).
- Hu, K. S., Yang, Y. Y., Wang, Y. X., Duan, X. G. & Wang, S. B. Catalytic carbon and hydrogen cycles in plastics chemistry. Chem. Catalysis 2, 724-761 (2022).
- Gao, Z. W., Ma, B., Chen, S., Tian, J. Q. & Zhao, C. Converting waste PET plastics into automobile fuels and antifreeze components. Nat. Commun. 13, 9 (2022).
- Pham, D. D. & Cho, J. Low-energy catalytic methanolysis of poly(ethyleneterephthalate). Green. Chem. 23, 511-525 (2021).
- Li, Y. W. et al. Catalytic transformation of PET and CO2 into high-value chemicals. Angew. Chem. Int. Ed. 61, 5 (2022).
- Barnard, E., Arias, J. J. R. & Thielemans, W. Chemolytic depolymerisation of PET: a review. Green. Chem. 23, 3765-3789 (2021).
- Zhang, S. B. et al. PET recycling under mild conditions via substituent-modulated intramolecular hydrolysis. Chem. Sci. 14, 6558-6563 (2023).
- Westhues, S., Idel, J. & Klankermayer, J. Molecular catalyst systems as key enablers for tailored polyesters and polycarbonate recycling concepts. Sci. Adv. 4, 8 (2018).
- Jing, Y. X. et al. Towards the circular economy: converting aromatic plastic waste back to arenes over a Ru/Nb2O5 catalyst. Angew. Chem. Int. Ed. 60, 5527-5535 (2021).
- Zhou, H. et al. Electrocatalytic upcycling of polyethylene terephthalate to commodity chemicals and H2 fuel. Nat. Commun. 12, 9 (2021).
- Lopez-Fonseca, R., Duque-Ingunza, I., de Rivas, B., Flores-Giraldo, L. & Gutierrez-Ortiz, J. I. Kinetics of catalytic glycolysis of PET wastes with sodium carbonate. Chem. Eng. J. 168, 312-320 (2011).
- Yang, W. S. et al. Easily recoverable and reusable p-toluenesulfonic acid for faster hydrolysis of waste polyethylene terephthalate. Green. Chem. 24, 1362-1372 (2022).
- Liu, Y. C. et al. Degradation of poly(ethylene terephthalate) catalyzed by metal-free choline-based ionic liquids. Green. Chem. 22, 3122-3131 (2020).
- Kang, M. J., Yu, H. J., Jegal, J., Kim, H. S. & Cha, H. G. Depolymerization of PET into terephthalic acid in neutral media catalyzed by the ZSM-5 acidic catalyst. Chem. Eng. J. 398, 9 (2020).
- Pinto, A. V. et al. Reaction mechanism of MHETase, a PET degrading enzyme. ACS Catal. 11, 10416-10428 (2021).
- Palm, G. J. et al. Structure of the plastic-degrading Ideonella sakaiensis MHETase bound to a substrate. Nat. Commun. 10, 10 (2019).
- Wang, C. Y. & El-Sepelgy, O. Reductive depolymerization of plastics catalyzed with transition metal complexes. Curr. Opin. Green. Sustain. Chem. 32, 6 (2021).
- Ye, M. X. et al. Ruthenium/TiO2-catalyzed hydrogenolysis of polyethylene terephthalate: reaction pathways dominated by coordination environment. Angew. Chem. Int. Ed. 135, e202301024 (2023).
- Li, X., Wang, J. Y., Zhang, T., Wang, T. F. & Zhao, Y. X. Photoelectrochemical catalysis of waste polyethylene terephthalate plastic to coproduce formic acid and hydrogen. ACS Sustain. Chem. Eng. 10, 9546-9552 (2022).
- Liu, F. L., Gao, X. T., Shi, R., Tse, E. C. M. & Chen, Y. A general electrochemical strategy for upcycling polyester plastics into added-value chemicals by a CuCo2O4 catalyst. Green. Chem. 24, 6571-6577 (2022).
- Wang, J. Y. et al. Electro-reforming polyethylene terephthalate plastic to co-produce valued chemicals and green hydrogen. J. Phys. Chem. Lett. 13, 622-627 (2022).
- Wang, J. Y. et al. Electrocatalytic valorization of poly(ethylene terephthalate) plastic and CO2 for simultaneous production of formic acid. ACS Catal. 12, 6722-6728 (2022).
- Iwai, Y., Gligorich, K. M. & Sigman, M. S. Aerobic alcohol oxidation coupled to palladium-catalyzed alkene hydroarylation with boronic esters. Angew. Chem. Int. Ed. 47, 3219-3222 (2008).
- Ranjani, G. & Nagarajan, R. Insight into copper catalysis: in situ formed nano Cu2O in Suzuki-Miyaura cross-coupling of aryl/indolyl boronates. Org. Lett. 19, 3974-3977 (2017).
- Li, R. H. et al. Thiophene-alkyne-based CMPs as highly selective regulators for oxidative Heck reaction. Org. Lett. 19, 4432-4435 (2017).
- Zhou, Y. B. et al. Conjugated microporous polymer as heterogeneous ligand for highly selective oxidative Heck reaction. J. Am. Chem. Soc. 139, 3966-3969 (2017).
- Yasuda, N. Application of cross-coupling reactions in Merck. J. Organomet. Chem. 653, 279-287 (2002).
- Cox, P. A. et al. Base-catalyzed aryl-B(OH)2 protodeboronation revisited: from concerted proton transfer to liberation of a transient aryl anion. J. Am. Chem. Soc. 139, 13156-13165 (2017).
- Hayes, H. L. D. et al. Protodeboronation of (hetero)arylboronic esters: direct versus prehydrolytic pathways and self-/auto-Catalysis. J. Am. Chem. Soc. 143, 14814-14826 (2021).
- Huang, W. W. et al. Highly efficient application of Mg/Al layered double oxides catalysts in the methanolysis of polycarbonate. Appl. Clay Sci. 202, 9 (2021).
- Nishimura, S., Takagaki, A. & Ebitani, K. Characterization, synthesis and catalysis of hydrotalcite-related materials for highly efficient materials transformations. Green. Chem. 15, 2026-2042 (2013).
- Hincapie, G., Lopez, D. & Moreno, A. Infrared analysis of methanol adsorption on mixed oxides derived from Mg/Al hydrotalcite catalysts for transesterification reactions. Catal. Today 302, 277-285 (2018).
- Gao, Y. S. et al. Comprehensive investigation of CO2 adsorption on Mg-Al-CO3 LDH-derived mixed metal oxides. J. Mater. Chem. A 1, 12782-12790 (2013).
- Di Serio, M. et al. Transesterification of soybean oil to biodiesel by using heterogeneous basic catalysts. Ind. Eng. Chem. Res. 45, 3009-3014 (2006).
- Shumaker, J. L. et al. Biodiesel synthesis using calcined layered double hydroxide catalysts. Appl. Catal. B: Environ. 82, 120-130 (2008).
- Abdellattif, M. H. & Mokhtar, M. MgAl-layered double hydroxide solid base catalysts for Henry reaction: a green protocol. Catalysts 8, 16 (2018).
- Montanari, T. et al. Zinc-aluminum hydrotalcites as precursors of basic catalysts: Preparation, characterization and study of the activation of methanol. Catal. Today 152, 104-109 (2010).
- Yan, B. H. et al. Green recycling of waste PET plastic monomers by banana peel extract. Chem. Eng. J. 474, 10 (2023).
- Zhao, Q., Tian, S. L., Yan, L. X., Zhang, Q. L. & Ning, P. Novel HCN sorbents based on layered double hydroxides: Sorption mechanism and performance. J. Hazard. Mater. 285, 250-258 (2015).
- Shangguan, E. et al. ZnAl-layered double hydroxide nanosheets-coated ZnO@C microspheres with improved cycling performance as advanced anode materials for zinc-based rechargeable batteries. J. Power Sources 422, 145-155 (2019).
- Yan, Q. H. et al. Promotional effect of Ce doping in Cu4Al1Ox-LDO catalyst for low-T practical NH3-SCR: Steady-state and transient kinetics studies. Appl. Catal. B: Environ. 255, 18 (2019).
Table 1. Relation of type I band area and DMT yield of PET depolymerization over Mg-Al-LDOs (the areas are integrated and normalized by catalyst weight).

Scheme 1 is available in the Supplementary Files section.
There is NO Competing Interest.
- SupplementaryInfo.pdf
Supplementary information
- Scheme1.png
Scheme 1. Strategy. Waste PET upcycling by in situ capturing EG with arylboronic acid in the methanolysis process.
{kind=link}