Differential Expression Profile and Screening of Disulfide Death-Related Genes
This figure (Fig. 1A) depicts the methodology used in our investigation. A notable disparity in the expression of 75 genes was observed between normal and glioma tissues (Fig. 1B). Furthermore, we conducted further refinement of the gene sets by selecting those with an absolute log2-fold change exceeding one, as seen in Fig. 1C. Subsequently, an investigation was conducted to analyze the variations in these genes between glioma and healthy tissues, as seen in Fig. 1D. In this study, a single-variate Cox regression model was employed to ascertain the correlation between twelve specific genes and disulfide death, as depicted in Fig. 1E.
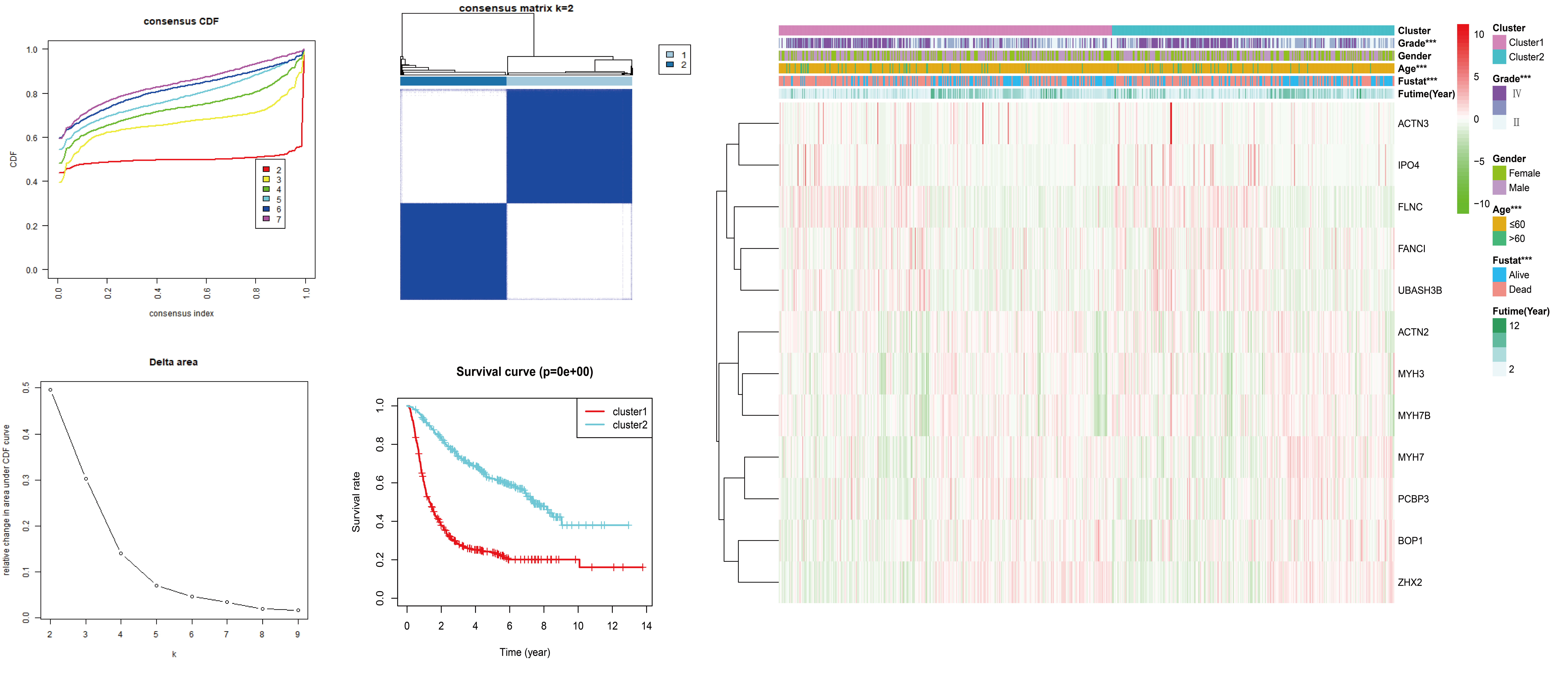
Identification of Glioma Clusters Using Consensus Clustering
A clustering analysis was performed on collection of 12 genes that are linked to disulfide mortality, with the aim of identifying different molecular groups. After considering the clinical significance and evaluating the CDF, it was found that value of k = 2 was the appropriate choice for further analysis of the clusters. The glioma samples were used to generate two distinct clusters, referred to as Cluster 1 and Cluster 2, as seen in Fig. 2A-C. Furthermore, it was noted that the individuals belonging to Cluster 1 had the most limited OS, as illustrated in Fig. 2D. Subsequently, a comparison was made between the molecular and clinical characteristics of these two groupings. There was significant correlation between age, living conditions, and several other characteristics and the World Health Organization grade in Cluster 1, as shown in Fig. 2E. Furthermore, the CGGA database was used to validate these results [Supplementary Figure S1].
Risk Signature for Glioma Constructed from 7 Selected Disulfide Death-Related Genes
Following the application of LASSO analysis to the set of twelve genes associated with disulfide death, a total of 7 genes (ACTN3, ACTN2, BOP1, IPO4, ZHX2, FLNC, and FANCI) were found to have significant associations with prognosis. These genes were then used to construct a risk model, as seen in Figs. 3A–B. In addition, it is important to highlight that the validation set, as depicted in Supplementary Figure S2, and the training set, as illustrated in Fig. 3C-D, exhibited an area under the AUC of ROC curves above the threshold of 70%. The training cohort was divided into two risk groups [Figure 3E] according to the median risk scores. It is noteworthy that patients classified as high-risk had lower OS rates, as shown in Fig. 3G, as well as shorter survival times, as depicted in Fig. 3F. In order to substantiate the accuracy of our model, it was observed that the high-risk group had a comparatively shorter duration of progression-free survival, as seen in Fig. 3H. Figure 3I demonstrates notable disparities between the high-risk and low-risk categories in relation to World Health Organization grade, patient age, living status, and living time. Furthermore, it provides a prediction of the probability of patient survival in future years based on factors such as gender, age, risk level, and grade [Figure 3J]. The findings of this study indicate that a risk profile, derived from the 7 specific genes linked to disulfide mortality, might possibly function as a predictive biomarker for assessing the prognosis of individuals with glioma.
Enrichment Analysis of Risk Features
To examine potential variations in the functional characteristics of the 7 disulfide death-related genes, functional enrichment analysis was conducted on the groups categorized by risk. The GO enrichment analysis revealed a notable increase in cellular localization associated with the malignant progression of gliomas, as well as a correlation between cell cycle regulation and cell proliferation, control of the NF-kB signaling pathway, and cell-matrix adhesion (Fig. 4A). Furthermore, the KEGG pathway analysis revealed that leukocyte transendothelial migration, cytoskeletal modification, and focal adhesion were significantly enriched (Fig. 4B). The results of this study indicate a correlation between the malignant biological mechanisms of glioma and the associated risk factors. The high-risk group was demonstrated to be strongly linked with apoptosis, Epithelial-Mesenchymal Transition (EMT), MTORC1-signaling, and the IL-2_STAT5 signaling pathway, according to GSEA analysis (Fig. 4C-F).
Tumor Microenvironment
The assessment of markers for the TME was performed for each sample using the ESTIMATE technique. Subsequently, a comparison of TME characteristics was made between the two risk groups. Based on the results shown in Fig. 5A, it may be seen that the high-risk group had elevated scores in stromal, immunologic, and ESTIMATE evaluations. The quantification of immune cells in the two groups at risk was assessed by the ssGSEA algorithm. as seen in Fig. 5B. The findings of the study revealed that the high-risk group had an increase in the presence of M2 macrophages, M1 macrophages, CD8 + T cells, and Tregs. On the other hand, the group with low risk demonstrated an increase in the presence of activated natural killer (NK) cells, monocytes, and activated mast cells. The possible impact of immune cell interactions, as seen in Fig. 5C, on the effectiveness of immunotherapy has the capacity to influence the selection of treatment options. The correlation found between resting NK cells and Tregs is of particular significance. Furthermore, the heatmap depicted the distinct activation patterns of immune pathways in the low- and high-risk groups. This observation indicated significant disparities in immune pathways, such as the Type I and II IFN response, inflammation-promoting pathways, checkpoint inhibition, and T-cell co-stimulation pathways, between the two risk groups (Fig. 5D). The present study has shown the capacity to forecast the cellular immunological characteristics of gliomas by leveraging risk factors.
Immune Checkpoints and Chemotherapy Drug Sensitivity
In addition, we performed an assessment of the correlation between risk factors and immune checkpoint genes. The findings of this study revealed that there was a significant upregulation of PD-1(programmed cell death protein 1), CTLA4(Cytotoxic T lymphocyte associate protein-4), PD-L1(Programmed Cell Death Ligand 1), CD28, CD80, and CD86 in the high-risk group. Conversely, the low-risk group exhibited overexpression of LDHB, LAMA3, VTCN1, JAK1, and IL12A (Fig. 6A). The genes TTN, TP53, and MUC16 exhibited a notable frequency of mutations, above 10%, in both the high-risk and low-risk groups (Fig. 6B–C). In both cohorts, the TP53 gene exhibited a significantly elevated mutation frequency, with rates of 36% and 50% observed, respectively. The group at high risk had a higher tumor mutation load in the research on mutations (Fig. 6D). The group at greater risk demonstrated a decrease in long-term survival rates as the tumor mutation load rose, as seen in Fig. 6E-F. With regard to the present utilization of chemotherapeutic agents in the management of gliomas, our investigation sought to evaluate the efficacy of these pharmacological therapies in two separate risk categories. It was determined that the high-risk population may exhibit increased vulnerability to commonly administered chemotherapy agents (Afatinib, cyclophosphamide, tamoxifen, Lapatinib, and Sorafenib; Fig. 6G-K), as shown by predictive models for three distinct chemotherapy treatments.
Silencing of IPO4 Inhibits Proliferation and Migration/Invasion of Glioma Cells
A research investigation was undertaken to better explore the involvement of 7 disulfide death-related genes in gliomas. Based on our first investigation, it is evident that the gene IPO4 in glioma cells has yet to receive much attention in terms of functional testing. Consequently, a decision was made to conduct a functional analysis of gliomas using IPO4. Initially, we conducted a comprehensive analysis of the TCGA and GTEx databases to confirm the higher expression of IPO4 in glioma tissues compared to normal brain tissues. We performed an extensive examination of the TCGA and GTEx databases to validate the elevated expression of IPO4 in glioma tissues relative to normal brain tissues. Following that, the technique of real-time PCR was employed to evaluate the comparative degree of IPO4 activity in cell lines associated with glioma (as seen in Fig. 7A). Compared to other glioma cell lines, the expression of IPO4 was notably higher in the U251, SF126, and U87 cell lines (Fig. 7B). RT-qPCR was used to verify the knockdown effectiveness. The obtained findings indicated that IPO4 sequences 1 and 2 exhibited higher knockdown efficiency in U251, SF126, and U87 cell lines, as shown in Fig. 7C. Following this, small interfering RNA (siRNA) was employed to promote the downregulation of IPO4 in U251, SF126, and U87 cell lines. The confirmation of suppression effectiveness was achieved using Western blotting analysis, as depicted in Fig. 7D. The experimental findings from the CCK8 experiment provided evidence that the silencing of IPO4 resulted in a decrease in cell viability, as seen in Fig. 7E–G. Both Cyclin D1 and PCNA, which are often used as markers for cell proliferation, exhibit a consistent decrease in their expression levels, as seen in Fig. 7H-I. Finally, the results obtained from transwell tests demonstrated that the migratory and invasive capacities of glioma cells were significantly impaired when IPO4 expression was suppressed, as seen in Fig. 8C-D. Notable modifications are also seen in the cell migration markers MMP9 and MMP10, as shown in Fig. 8A-B. The results imply that IPO4 holds promise as a potential candidate for therapeutic interventions in the management of glioma.
{kind=link}
{kind=link}