3.1 Synthesis and X-Ray Crystal Study
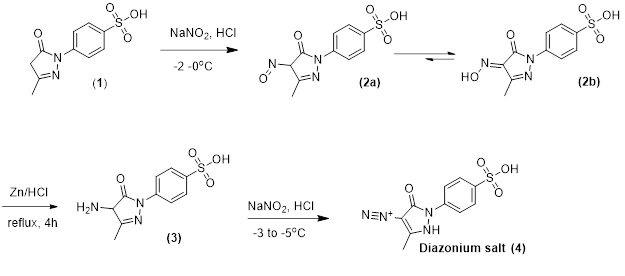
Synthesis of diazonium salt of (4) was achieved according to the route sketched in Scheme 1.
1-(4-sulphophenyl) 3-methyl-2-pyrazolin-5-one (1) was nitrosated at -2-0 oC using NaNO2 and HCl as described by Knorr1 to afford the nitroso compound (2) which was filtered to remove some terry material. The nitroso derivative (2a), usually exists in its tautomeric oxime (2b) form as indicated by its FTIR spectrum, was salted out by common salt, and dried after filtration. Reduction of (2) was achieved and zinc metal in the presence of HCl added in small portions at reflux to afford a colorless solution. A small amount of additional zinc was added, and the resultant amine hydrochloride was quenched to -7 ºC. The excessive un-reacted zinc was removed by filtration. The amine hydrochloride (3) was diazotized very carefully by using an aqueous solution of NaNO2 and HCl at -5 to -3 ºC to provide the title compound (4). The careful control of temperature is essential to avoid the formation of rubazoic acid, which is routinely formed during this reaction on increasing temperature due to oxidizing action of nitrous acid formed in situ.
In the molecular structure of diazonium salt the phenyl- as well as the pyrazolone-rings lie almost in plane, the relevant torsion angle C1-N1-C5-C6 measures 3.3(5)°. Essential bonding parameters of the pyrazolone moiety are N1—N2 [1.407(3) Å], C1—O1 [1.230(3) Å], C2—N3 [1.325(4) Å], N3—N4 [1.110(3) Å] and C2—N3—N4 [177.3(3)°]. These are in close agreement with those of 4-Diazonio-2-methyl-5-nitro-3-oxo-2,3-dihydropyrazol-1-ide (WETGEJ)x1with N—N [1.362 Å], C—N2 [1.323 Å], (C)N—N [1.116 Å] and C—N—N [177.7o].
The crystal structure (Fig. 2) shows various hydrogen bonding patterns with the solvent water molecules. Strongest interactions are O10—H11···O11iii [symmetry code: (iii) -x + 1, -y + 1, -z + 2] with H···O of 1.94(1) Å, O20—H21···O12iv [symmetry code: (iv) x, y + 1, z] with 1.98(1) Å, O10—H12···O12i [symmetry code: (i) x-1, y + 1, z] with 2.01(2) Å and O20—H22···N2 with 2.14(1) Å. An intramolecular C6—H6A···O1 bond is connected with the planar arrangement of both the aromatic ring planes.
The asymmetric unit of diazonium salt is depicted in Fig. 1; the unit cell with intermolecular H-bonding pattern is shown in Fig. 2 (Table 2). Table 3 gives the selected bond lengths (Å) and angles (°).
Table 2
Hydrogen-bond geometry (Å, º).
|
D—H···A
|
D—H
|
H···A
|
D···A
|
D—H···A
|
|
C6—H6A···O1
|
0.95
|
2.21
|
2.878 (4)
|
126
|
|
O10—H12···O12i
|
0.84 (1)
|
2.01 (2)
|
2.808 (3)
|
161 (3)
|
|
O10—H11···O11iii
|
0.84 (1)
|
1.94 (1)
|
2.774 (3)
|
173 (4)
|
|
O20—H22···N2
|
0.85 (1)
|
2.14 (1)
|
2.981 (4)
|
173 (4)
|
|
O20—H21···O12iv
|
0.84 (1)
|
1.98 (1)
|
2.810 (4)
|
172 (5)
|
| Symmetry codes (i) x − 1, y + 1, z; (iii) − x + 1, −y + 1, −z + 2; (iv) x, y + 1, z. |
Table 3
Selected geometric parameters (Å) of 1-(p-sulphophenyl)-3-methyl-4-azo-5- pyrazolone.
|
Na—O1i
|
2.2763 (15)
|
N1—C6
|
1.417 (2)
|
|
Na—O5
|
2.2907 (17)
|
N2—C9
|
1.303 (2)
|
|
Na—O6
|
2.3630 (16)
|
N3—N4
|
1.109 (2)
|
|
Na—O4
|
2.3733 (15)
|
N3—C8
|
1.328 (2)
|
|
Na—O6ii
|
2.4951 (16)
|
C1—C2
|
1.382 (3)
|
|
Na—S1
|
3.3338 (9)
|
C1—C6
|
1.387 (3)
|
|
Na—Naii
|
3.6887 (14)
|
C1—H1
|
0.9300
|
|
S1—O2
|
1.4387 (16)
|
C2—C3
|
1.380 (3)
|
|
S1—O4
|
1.4439 (14)
|
C2—H2
|
0.9300
|
|
S1—O3
|
1.4543 (16)
|
C3—C4
|
1.385 (3)
|
|
S1—C3
|
1.7720 (18)
|
C4—C5
|
1.383 (3)
|
|
O1—C7
|
1.225 (2)
|
C4—H4
|
0.9300
|
|
O1—Naiii
|
2.2763 (15)
|
C5—C6
|
1.386 (2)
|
|
O5—H1
|
0.79 (3)
|
C5—H5
|
0.9300
|
|
O5—H2
|
0.75 (3)
|
C7—C8
|
1.433 (2)
|
|
O6—Naii
|
2.4951 (16)
|
C8—C9
|
1.413 (3)
|
|
O6—H3
|
0.80 (3)
|
C9—C10
|
1.488 (3)
|
|
O6—H4
|
0.84 (3)
|
C10—H10A
|
0.9600
|
|
N1—C7
|
1.385 (2)
|
C10—H10B
|
0.9600
|
|
N1—N2
|
1.405 (2)
|
C10—H10C
|
0.9600
|
| Symmetry codes: (i) x-1, y, z-1; (ii) -x-1, -y + 1, -z; (iii) x + 1, y, z + 1. |
3.2 Hirshfeld surface analysis
To visualize the intermolecular interactions in the crystal of the title compound, a Hirshfeld surface (HS) analysis [27] was carried out by using Crystal Explorer 17.5 [28]. In the HS plotted over dnorm (Fig. 3), the white surface indicates contacts with distances equal to the sum of van der Waals radii, and the red and blue colours indicate distances shorter (in close contact) or longer (distinct contact) than the van der Waals radii, respectively [29]. The bright-red spots appearing near Na, N2, O11, O12 and hydrogen atoms H11, H12, H21, H22 indicate their roles as the respective donors and/or acceptors; they also appear as blue and red regions corresponding to positive and negative potentials on the HS mapped over electrostatic potential [30] as shown in Fig. 4. The blue regions indicate positive electrostatic potential (hydrogen-bond donors), while the red regions indicate negative electrostatic potential (hydrogen-bond acceptors). The shape-index of the HS is a tool to visualize the π ... π stacking by the presence of adjacent red and blue triangles; if there are no adjacent red and/or blue triangles, then there are no π ... π interactions. Figure 5 clearly suggests that there are no π ... π interactions in (I). The overall two-dimensional fingerprint plot, Fig. 6a, and those delineated into H ··· H, H ··· O/O ··· H, H ··· N/N ··· H, O ··· NA/NA ··· O, N ··· O/O ··· N, C ··· C, C ··· N/N ··· C, H ··· C/C ··· H, N ··· N, H ··· NA/NA ··· H, C ··· O/O ··· C, O ··· O, N ··· NA/NA ··· N and H ··· S/S ··· H [31] are illustrated in Figs. 4b—o, respectively, together with their relative contributions to the Hirshfeld surface. The most important interaction is H ··· H (Table 4) contributing 27.5% to the overall crystal packing, which is reflected in Fig. 6b as widely scattered points of high density due to the large hydrogen content of the molecule with the tip at de = di = 1.08 Å. The pair of spikes in the fingerprint plot delineated into H ··· O/O ··· H contacts (Table 4) have a symmetrical distribution of points (26.6% contribution, Fig. 6c) with the tips at de + di = 1.78 Å. The pair of characteristic wings resulting in the fingerprint plots delineated into H ··· N/N ··· H, Fig. 6d, contacts with 8.9% contribution to the HS arises from the H ··· N/N ··· H contacts (Table 4) and is viewed as pair of spikes with the tips at at de + di = 1.96 Å. The O ··· NA/NA ··· O contacts (8.1%, Fig. 6e) have a scissor-shaped distribution of points with the tips at de + di = 2.31 Å. The N ··· O/O ··· N contacts (7.5%, Fig. 6f) have a fringed-shaped distribution of points with the tips at de + di = 2.79 Å and de + di = 2.82 Å for inner and outer fringes, respectively. The C ··· C contacts (6.6%, Fig. 6g) (Table 4) have a bullet-shaped distribution of points with the tip at de = di = 1.72 Å. The C ··· N/N ··· C contacts (Table 4) contribute 5.9% to the overall crystal packing, which are reflected in Fig. 6h with the small tips at de + di = 3.28 Å. In the absence of C—H ··· π interactions, the pair of characteristic wings in the fingerprint plot delineated into H ··· C/C ··· H (Table 4) contacts (Fig. 6i, 3.2% contribution to the HS) has the tips at de + di = 2.96 Å. The N ··· N contacts (Table 4) contribute 2.2% to the overall crystal packing, which are reflected in Fig. 6j with the tip at de = di = 1.46 Å. The H ··· NA/NA ··· H contacts have an unsymmetrical distribution of points (1.6% contribution, Fig. 6k) with the tips at de + di = 3.28 Å. Finally, the C ··· O/O ··· C (0.7%, Fig. 6l), O ··· O (0.7%, Fig. 6m), N ··· NA/NA ··· N (0.5%, Fig. 6n) and H ··· S/S ··· H (0.1%, Fig. 6o) contacts with contribution smaller than 1.0% to the HS have scattered points of very low density.
The Hirshfeld surface representations with the function dnorm plotted onto the surface are shown for the H ··· H, H ··· O/O ··· H, H ···N/N ··· H and O ··· NA/NA ··· O, interactions in Figs. 7a—d, respectively.
The Hirshfeld surface analysis confirms the importance of H-atom contacts in establishing the packing. The large number of H ··· H and H ··· O/O ··· H interactions suggest that van der Waals interactions and hydrogen bonding play the major roles in the crystal packing [32].
Table 4
Selected interatomic distances (Å).
|
H12···S1i
|
2.90 (4)
|
O20···H10A
|
2.87
|
|
S1···H4Cii
|
3.1624
|
N2···C7ii
|
3.446 (4)
|
|
S1···H11iii
|
3.13 (4)
|
N3···C9v
|
3.323 (4)
|
|
H21···S1iv
|
3.02 (5)
|
N3···N4ix
|
3.091 (4)
|
|
O1···N3
|
2.948 (4)
|
N4···C9v
|
3.437 (4)
|
|
O1···C6
|
2.878 (4)
|
N4···N4ix
|
2.950 (4)
|
|
O10···O11iii
|
2.774 (3)
|
H22···N2
|
2.14 (3)
|
|
O11···N4v
|
2.814 (4)
|
N2···H10A
|
2.46
|
|
O20···O12iv
|
2.810 (4)
|
H9A···N4vi
|
2.66
|
|
O13···N4ii
|
3.016 (4)
|
C1···C10v
|
3.543 (4)
|
|
O13···C2ii
|
3.259 (4)
|
C1···C5v
|
3.589 (4)
|
|
O13···N3ii
|
2.873 (4)
|
C1···C9ii
|
3.585 (4)
|
|
O13···N4vi
|
3.117 (4)
|
C2···C10v
|
3.500 (4)
|
|
O20···N2
|
2.981 (4)
|
C2···C5v
|
3.585 (4)
|
|
O1···H6A
|
2.21
|
C3···C6v
|
3.555 (5)
|
|
O11···H11iii
|
1.93 (4)
|
C3···C7ii
|
3.568 (4)
|
|
O11···H9Avii
|
2.79
|
C3···C8ii
|
3.559 (4)
|
|
O12···H7A
|
2.45
|
C5···C5ii
|
3.498 (4)
|
|
H12···O12i
|
2.01 (4)
|
C1···H6A
|
2.66
|
|
H21···O12iv
|
1.98 (4)
|
C3···H22
|
2.91 (3)
|
|
O12···H4Cii
|
2.66
|
H4A···H22
|
2.47
|
|
O20···H4Bviii
|
2.85
|
H10A···H22
|
2.35
|
|
O20···H4A
|
2.84
|
|
|
Symmetry codes: (i) x − 1, y + 1, z; (ii) − x + 2, −y + 1, −z + 1; (iii) − x + 1, −y + 1, −z + 2; (iv) x, y + 1, z; (v) − x + 1, −y + 1, z + 1; (vi) x + 1, y, z + 1; (vii) − x + 2, −y + 1, −z + 2; (viii) − x + 1, −y + 2, −z + 1; (ix) − x + 1, −y + 1, −z.
3.3 Optical and Electrochemical Studies
The optical and electrochemical studies of newly synthesized diazonium salt were conducted to get information regarding the absorption intensity and redox behavior.
The UV-visible absorption spectrum of diazonium salt of 4-amino-p-sulphophenyl-3 -methyl − 5-pyrazolone in ethanol is shown in Fig. 8. The absorption maximum (λmax) of diazonium was taken in ethanol (1x10− 5 M solution) to observe the molar extinction coefficient of the compound. The molar extinction was found to be log ɛ = 6.5 and λmax of diazonium was found at 420 nm. This transition can be assigned as due to n-π* and π-π* transitions of the azo linkage. This value of the molar extinction coefficient is the evidence of the high absorption intensity. These transitions are bathochromic shifted to higher λmax due to the presence of o-hydroxy group to diazo linkage. The hydroxy group has the ability to donate the lone pair of electrons to ring which increases the electron density at the chromophoric motif and hence decreases the energy required for electronic transitions. Moreover, this compound undergoes tautomerism and keto form is formed which increases the stability of azo linkage through strengthening the carbon to nitrogen atom of azo linkage.
The electrochemical characterization of diazonium salt was made by cyclic voltammetry using DMSO containing 0.1 M TBAPF6 as a supporting electrolyte with glassy carbon electrode. All redox potentials, HOMO (highest occupied molecular orbital), LUMO (lowest unoccupied molecular orbital) and band gap energies (Eg) were calculated from this technique.
The reduction potential of diazonium compound was observed from cyclic voltammogram (Fig. 9) and was found to be of valued 0.9 V. By utilizing the reduction potential of the compound, the HOMO and LUMO levels were determined which were found at levels of -6.45 and − 3.5 eV, respectively, with the help of Bredas Equations (Eqs. 1, 2 and 3). This diazonium has a high electron affinity (LUMO level − 3.5 eV) and may act as an acceptor material for organic heterojunction solar cells. The band gap energy (Eg) of the compound was 2.95 eV (Table 5). This band gap energy is moderate which is affected by the o-hydroxyl group. This OH group donates electron to pyrazole ring and pyrazole is formed which leads to extension of conjugation and decreasing the energy of LUMO levels too much which reduces the gap between the two.
Eq. 1 ELUMO= - (4.4 + Ered)
Eq. 2 EHOMO= ELUMO -Eg
Eq. 3 Eg= 1240 eV nm/ λ
Where,
E HOMO = energy of HOMO level (eV)
E LUMO = energy of LUMO level (eV)
E g = band gap energy (eV)
E red = half-wave potential (V)
λ = cut-off wavelength of the absorption band (nm)
Table 5
Band gap energy (Eg), HOMO and LUMO levels of 1-(p-sulphophenyl)-3-methyl-4-azo-5- pyrazolone.
|
Compound
|
Eg (eV)
|
LUMO (eV)
|
HOMO (eV)
|
|
Diazonum Salt
|
2.95
|
-3.5
|
-6.45
|
{kind=link}
{kind=link}
{kind=link}