Human faecal samples collection
Study participants were recruited between 2017 and 2022 from Kyoto University (permit number: R2875-4), Keio University (permit number: 20210021), Kobe University (permit number: B210124), Kyoto Medical Center (permit number: 20–074), Tokyo University of Agriculture and Technology (permit number: 210704-2846) and Fukujuji Hospital (permit number: 21016). The volunteers were Japanese individuals aged 20–80 years. The exclusion criteria were as follows: Participants with a BMI below 18.5 or above 60 kg m− 2; those who regularly took medication with proton pump inhibitors; those with diabetes and hyperlipidemia; those who used antibiotics within 2 weeks; and those who consumed probiotic supplements, including milk, yogurt, and fermented food before sample collection. All the participants involved in this study provided written informed consent. Faecal samples were collected using a stool collection tube and stored at − 80°C until preparation and analysis.
Faecal samples cultured condition
Human and mouse faecal samples were cultured on MRS agar (Difco Laboratories Inc., Detroit, MI, USA) or MRS agar containing 15% fructose, galactose, glucose, lactose, maltose, and sucrose at 30°C for 48 h under anaerobic conditions. EPS product colonies were picked and underwent 16S ribosomal RNA (rRNA) gene amplification using the primers 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-GGTTACCTTGTTACGACTT-3′). The PCR products were purified using an UltraClean PCR Clean-Up Kit (MO BIO Laboratories, San Diego, CA, USA), and directly sequenced using a Big Dye Terminator Cycle Sequencing Kit ver. 3.1 (Applied Biosystems, Foster City, CA, USA) and an ABI 3730xl DNA analyzer system (Applied Biosystems). The isolated strains shared more than 98% similarity in their 16S rRNA gene sequences.
Bacterial culture
The cultivation of S. salivarius in MRS medium containing 15% sucrose, 15% glucose, 15% fructose, and 7.5% glucose + 7.5% fructose was monitored for 24 h. The dominant gut bacteria were selected using a human gut microbial gene catalog25 obtained from the Japan Collection of Microorganisms (JCM). Bacteria were recovered according to the manufacturer’s instructions as previously described18. Intestinal bacteria were collected in nutrient broth (Difco Laboratories Inc.) containing 10% glycerol and stored at − 80°C.
Characterisation of S. salivarius-produced EPS
S. salivarius was cultured on MRS agar alone at 37°C or MRS agar containing 15% sucrose at 30°C for 48 h under anaerobic conditions and imaged using scanning electron microscopy (SEM; JSM-7500F; HUSRI, Aichi, Japan). SsEPS were collected from the agar plate and purified using ethanol precipitation, as previously described18 or dialysis membranes with a molecular cutoff of 3,500 Da (Snake Skin dialysis tubing, Thermo Fisher Scientific, Waltham, MA, USA). The precipitated SsEPS was dried over calcium chloride for 24–48 h. To determine its monosaccharide composition, SsEPS was extracted as described previously18, with certain modifications. Briefly, SsEPS was hydrolysed by addition of trifluoroacetic acid (0.5 M) and incubated at 120°C for 0.5–2 h. After incubation, the supernatant was filtered through a 0.45 µm filter. The monosaccharide composition was analysed by ligand exchange chromatography using an 8.0 × 300 mm SUGAR SC1011 column (Shodex, Tokyo, Japan). Detection was performed using a RID-20A (Shimadzu, Kyoto, Japan), with D-glucose and D-fructose (Nacalai Tesque, Kyoto, Japan) as standards. The average molecular weight of SsEPS was determined by size exclusion chromatography using an 8.0 × 300 mm OHpak SB-800 HQ series column (Shodex). Standards for purchased pullulans (Shodex) and dextrans (Sigma-Aldrich, St. Louis, MO, USA) with average molecular weights of 1,600,000–5,900 and 1,500,000–2,800,000 Da, respectively, were established using calibration curves.
Structure of S. salivarius-produced EPS
The structure of the SsEPS was confirmed using 1H and 13C NMR spectroscopy. SsEPS was dissolved in 750 µL of D2O containing 0.1% 3-(trimethylsilyl) propionic-2,2,3,3-d4 acid sodium salt (TMSP). After allowing the solution to stand for 12 h, the 1H NMR spectrum was recorded using a JEOL ECA-500 spectrometer with a frequency of 500 MHz at 25°C. Chemical shifts are reported in δ (ppm) relative to TMSP as the chemical shift internal standard. 13C NMR spectra were recorded on a JEOL ECA-500 spectrometer with a frequency of 125 MHz at 25°C and are reported relative to TMSP signal as the chemical shift internal standard. The infrared (IR) spectra were recorded using a JASCO FT/IR-4100 spectrometer.
S. salivarius-produced levan: IR (neat cm− 1): 3415 (OH); 1H NMR (500 MHz, D2O): δ 4.20 (d, J = 8.0 Hz, 1H), 4.14–4.08 (m, 1H), 3.98–3.87 (m, 2H), 3.78 (d, J = 12.0 Hz, 1H), 3.69 (d, J = 12.0 Hz, 1H), 3.59–3.55 (m, 1H); 13C{1H} NMR (125 MHz, D2O): δ 107.1, 83.2, 79.2, 78.1, 66.2, 62.8.
S. salivarius-produced glucan: IR (neat cm− 1): 3375 (OH); 1H NMR (500 MHz, D2O): δ 4.99 (d, J = 2.9 Hz, 1H), 4.04–3.92 (m, 2H), 3.80–3.70 (m, 2H), 3.59 (dd, J = 9.7, 2.9 Hz, 1H); 13C{1H} NMR (125 MHz, D2O): δ 100.5, 76.2, 74.3, 73.0, 72.4, 68.4.
RNA isolation and quantitative reverse transcriptase (qRT)-PCR
S. salivarius was cultured in MRS medium containing 15% sucrose or 15% glucose at 30°C for 10 h under anaerobic conditions. Total RNA was extracted using the NucleoSpin RNA kit (Takara Bio, Shiga, Japan) and reverse-transcribed into cDNA using Moloney murine leukemia virus reverse transcriptase (Thermo Fisher Scientific). SYBR Premix Ex Taq II (Takara Bio) and StepOnePlus real-time PCR system (Applied Biosystems) were used for qRT-PCR analysis, as previously described18. SsEPS-synthesised enzyme primer sequences are listed in Extended Data Table 1.
RNA-sequencing data analysis
Sequencing libraries were constructed using the NEBNext rRNA Depletion Kit (Bacteria) (New England Biolabs, Inc., MA, USA) and the TruSeq Stranded mRNA Library Prep Kit (Illumina, CA, USA) according to the manufacturer's protocols. The sequencing libraries were sequenced on an Illumina HiSeq 2500 platform with 100 bp paired-end reads. On average, 1.2 million read pairs per sample were sequenced across eight samples (4 glucose samples and 4 sucrose samples). RNA-Seq data were analysed using the CLC Genomics Workbench (Qiagen Bioinformatics, Venlo, Netherlands) to identify differentially expressed genes. To obtain clean reads, low-quality reads were removed by trimming, whereas high-quality reads were aligned to the S. salivarius NCTC 7366 genome retrieved from the NCBI database. The parameters were set as follows: minimum length fraction = 0.8 and minimum similarity fraction = 0.8. Expression values were established as transcripts per million reads (TPM). The KEGG Pathway enrichment analysis was performed from the GhostKOALA result of expressed genes. The enriched pathway for the experiment was identified from the Welch's t-test result with false discovery rate correction (q < 0.01) using the R software environment. A gene set enrichment analysis was performed using the Kyoto Encyclopedia of Genes and Genomes database (KEGG) (http://www.genome.jp/kegg/).
Shotgun metagenomic sequencing data analysis
DNA was quantitated using Qubit fluorometric quantitation (Thermo Fisher Scientific) and qualified by DNA size profiling on a fragment analyzer (Agilent, Santa Clara, CA, USA). High molecular weight DNA (> 10 kbp; 3 µg) was used to build the library. DNA shearing into fragments of approximately 150 bp was performed using an ultrasonicator (Covaris, Woburn, MA, USA), and the DNA fragment library was constructed using the Ion Plus Fragment Library and Ion Xpress Barcode Adapters kits (Thermo Fisher). Purified and amplified DNA fragment libraries were sequenced using DNBSEQ-G400 (MGI Tech) with a minimum of 20 million high-quality reads of 150 bp (on average) generated per library. The paired-end sequences were merged by BBmaps (v38.84-0)34. They underwent preprocessing by Kneaddata (v0.12.0) to remove the host genome based on the human (hg37 dec_v0.1) and mouse (C57BL_6NJ) genome databases. The Whole genome sequence based axonomy profile was generated by MetaPhlAn (v4.0.4)35. Microbial gene families and metabolic pathways were assessed using HUMAnN3 (v3.8)36 based on the UniRef90 EC filtered database (uniref90_201901). MaAsLin2 was used to identify significant pathway from HUMAnN3 outputs37. All computational scripts are available on GitHub [https://github.com/petadimensionlab/EPS].
SCFAs measurement
SCFA levels in human faeces, murine faeces, and murine plasma were measured following a previously described modified protocol38. Ether layers containing SCFAs were collected and pooled for gas chromatography-mass spectrometry (GC-MS) using a GCMS-QP2010 Ultra GC mass spectrometer (Shimadzu). The SCFA concentration was evaluated over a specified concentration range.
EPS measurement.
The faecal contents (300 mg) were immediately mixed with five volumes of sterile distilled water containing 2% 5-sulfosalicylic acid and vortexed. The mixture was then centrifuged, and the supernatant containing the EPS was collected. Two volumes of hexane were added to the supernatant, which was then vortexed for 5 min. After centrifugation of the samples at 10,000 × g for 15 min, the water layers containing EPS were collected and subjected to HPLC analysis using an RID-20A (Shimadzu) and an 8.0 × 300 mm OHpak SB-800 HQ series column (Shodex).
Animal Study
C57BL/6J, Gpr41Gpr43 double-deficient, and ICR mice were housed under a 12-h light-dark cycle and fed normal chow (CE-2; CLEA, Tokyo, Japan). GF-ICR mice were housed in vinyl isolators under a 12-h light–dark cycle and fed normal chow (CL-2, 50kGy irradiated; CLEA). Gpr41Gpr43 double-deficient mice were generated as described previously12. All experimental procedures involving mice were performed according to the protocols approved by the Committee on the Ethics of Animal Experiments of the Kyoto University Animal Experimentation Committee (Lif-K21020) and Tokyo University of Agriculture and Technology (permit number: R05-47 and R05-48).
Four-week-old C57BL/6J and Gpr41Gpr43 double-deficient mice were fed a modified D12492 diet (60% kcal fat; Research Diets, New Brunswick, NJ, USA) for 12 weeks in high fat diet (HFD) studies. The composition of the modified diet is shown in Extended Data Table 2.
After fasting 24 h, 7-week-old C57BL/6J, Gpr41Gpr43 double-deficient, conventional ICR, and GF-ICR healthy male mice were fed 0.2 g AIN-93G, containing 50% cellulose or 50% SsEPS. After 1 h, glucose (2 g/kg body weight) was intraperitoneally administered to each mouse. Blood glucose levels in the tail vein were measured using a OneTouch UltraVue glucometer (LifeScan, Milpitas, CA, USA) and an LFS Quick Sensor (LifeScan) before and at 15, 30, 60, 90, and 120 min after injection. Plasma samples were collected from the inferior vena cava at 15 min after glucose administration for insulin and GLP-1 measurement18,39.
For the gnotobiotic experiments, 5-week-old GF-ICR mice were fed an AIN-93G diet (50kGy irradiated; Research Diets) for 4 weeks. After 2 weeks, each bacterial strain (1 × 108 CFU/mouse) was administered via oral gavage three times per week. Sterilised water containing 20% sucrose with or without 0.5% acarbose as an α-glucosidase inhibitor (Tokyo Chemical Industries, Japan), glucose, and fructose were administered for 2 weeks (fig. S7A and S7G). For long-term treatment, 7-week-old GF-ICR mice were fed an AIN-93G diet, D12492 diet (Research Diets), or modified D12492 diet (50kGy irradiated) for 9 weeks. Each bacterial strain (1 × 108 CFU/mouse) was administered via oral gavage three times a week at 7 and 11 weeks old. The composition of the modified D12492 diet is shown in Extended Data Table 3.
Faecal transplantation in animal experiment
The faecal samples from two women, Ss (+) (aged 43 with a BMI of 30.0 kg m− 2) and Ss (-) (aged 45 with a BMI of 40.5 kg m− 2) were suspended in equal volumes of nutrient broth (Difco Laboratories Inc.) containing 10% glycerol and stored at − 80℃ until use. The thawed frozen samples were cultured anaerobically at 37℃ for 24 hours in GAM medium (Nissui, Tokyo, Japan), filtered through a membrane paper, and orally inoculated into germ-free mice (approximately 250 µl per mouse). Faecal culture solutions were administered once weekly until 16 weeks of age.
Biochemical analyses
Blood glucose levels were measured using a OneTouch UltraVue glucometer (LifeScan) and an LFS Quick Sensor (LifeScan). The levels of plasma non-esterified fatty acids (LabAssayTM NEFA; Wako Pure Chemical Co. Ltd., Osaka, Japan), triglycerides (LabAssayTM Triglyceride; Wako Pure Chemical Co. Ltd.), total cholesterol (LabAssayTM Cholesterol; Wako Pure Chemical Co. Ltd.), insulin (Mouse Insulin enzyme-linked immunosorbent assay [ELISA]; Shibayagi, Gunma, Japan), and active glucagon like peptide-1 (GLP-1) (GLP-1 [Active] ELISA; Merck Millipore, Billerica, MA, USA) were measured according to the manufacturer’s instructions. To prevent degradation of active GLP-1, plasma samples were treated with a dipeptidyl peptidase IV inhibitor (Merck Millipore).
DNA extraction and gut microbial composition
DNA was extracted from faecal samples using the FastDNA SPIN kit for feces (MP Biomedicals, Irvine, CA, USA) as described previously18. Partial 16S rRNA gene sequences were amplified by targeting the hypervariable regions v4 using the primers 515F; 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGGTGYCAGCMGCCGCGGTAA-3′ and 806R; 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGGACTACHVGGGTWTCTAAT-3′. Amplicons generated from each sample were purified using AMPure XP Beads (Beckman Coulter, Brea, CA, USA), and appended with Nextera XT index kit (Illumina, San Diego, CA, USA). Amplicons were sequenced using a MiSeq sequencer (Illumina) and MiSeq Reagent kit (version 3.0; 600 cycles). The 16S rRNA sequence data were then processed using the quantitative insights into the microbial ecology 2 (QIIME 2) pipeline, and analysed using the MiSeq Reporter software with the SILVA database (Illumina). Diversity was analysed using QIIME script core_diversity_analyses.py. Permutational multivariate analysis of variance (QIIME script compare_categories.py) was used to assess the statistical significance of sample groupings. For quantitative PCR, SYBR Premix Ex Taq II (Takara Bio) and StepOnePlus real-time PCR system (Applied Biosystems) were used. The bacterial primer sequences are listed in Extended Data Tables 4 and 5.
Statistical analysis
The mean ± standard error of the mean is presented for all values. We assessed the normality of the data using the Shapiro–Wilk test (normal distribution was defined at p ≥ 0.05). To determine the statistical significance between two groups with normal distribution, we used Student's t-test. For groups with non-normal distribution, the Mann–Whitney U test was used for comparison. One-way analysis of variance (ANOVA) was used to compare data from multiple groups (three or more). For normally distributed sample sets, Dunnett’s post-hoc test was used, whereas the Kruskal–Wallis test paired with Dunn’s post-hoc test was used for non-normally distributed sample sets. Statistical significance was set at p < 0.05. Additionally, The Benjamini–Hochberg procedure was used to estimate the false discovery rates (Q-values) of the 16S rRNA gene sequencing data. This study analysed the correlations between microbiota and gut environmental factors. To calculate correlations, we used Spearman's rank correlation coefficients for bacterial genus abundance, including Muribaculum, Paramuribaculum, Duncaniella, Bacteroides, Akkermansia, Faecalitalea, Desulfovibrio, Streptococcus, Blautia, and Ruminococcus and faecal SCFAs, such as acetate, propionate, and n-butyrate. We selected only correlations with an absolute value above 0.6 and a Q-value below 0.05. Outliers were evaluated using the Smirnov–Grubbs test.
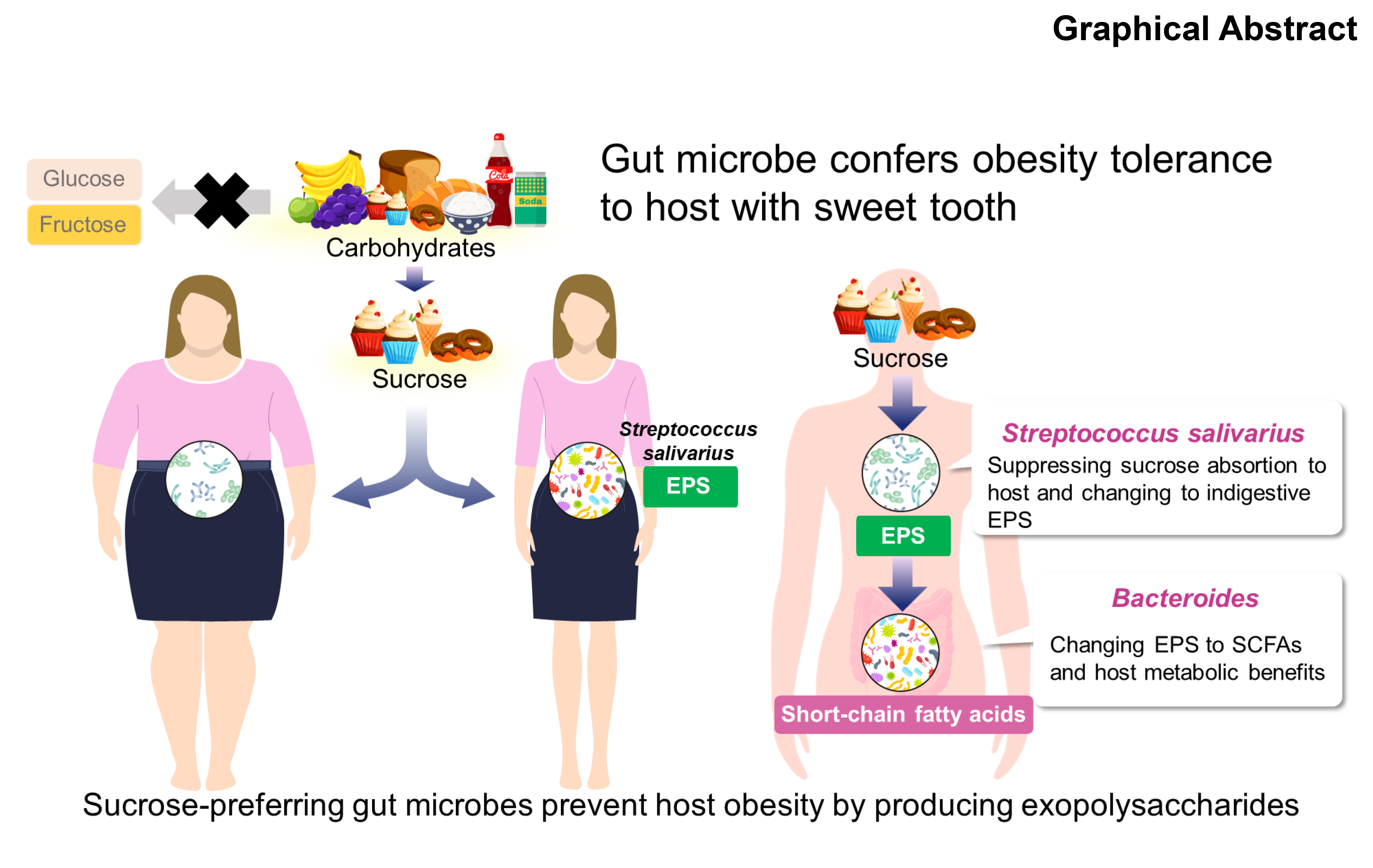{kind=link}