3.3.1 Optimized geometry
1, 3-Dimethyl-3H-benzimidazol-1-ium [DBZ] (a), 1, 3-Dimethyl-3H-benzimidazol-1-ium, monohydrate [DBZW] (b) 1, 3-Dimethyl-3H-benzimidazol-1-ium, iodine [DBZI] (c) and 1, 3-dimethyl-3H-benzimidazol-1-ium iodide monohydrate [DBZIW] (d) structures were optimized at the B3LYP method with 6-311G (d, p) level using Gaussain-09 software as shown in Fig. 8. By optimizing the benzimidazole derivatives both with and without iodine and water, it was possible to get to the conclusion regarding the involvement of these molecules in the benzimidazole derivatives. Table 3 compares the computed bond lengths and bond angles of (a) DBZ, (b) DBZW, (c) DBZI, and (d) DBZIW with experimental X-ray data on bond lengths and bond angles.
Table 3
Bond lengths (Å) and bond angles (°) involving non-hydrogen atoms based on X-ray data and computational calculations at the B3LYP /6-311G (d, p) level of theory (with estimated standard deviation in brackets).
| Bond lengths (Å) | Experimental X-ray | Theoretical calculations at B3LYP /6-311G (d, p) level of theory |
| DBZIW | DBZ | DBZW | DBZI | DBZIW |
| N1- C1 | 1.330(4) | 1.3928 | 1.4058 | 1.3709 | 1.3723 |
| N1- C2 | 1.461(4) | 1.4429 | 1.4475 | 1.4654 | 1.4334 |
| N1- C5 | 1.392(4) | 1.3843 | 1.3765 | 1.4098 | 1.4158 |
| N2- C1 | 1.331(4) | 1.3939 | 1.4099 | 1.3808 | 1.4158 |
| N2- C3 | 1.463(4) | 1.4421 | 1.4458 | 1.4653 | 1.4311 |
| N2- C4 | 1.392(4) | 1.3807 | 1.3821 | 1.4098 | 1.4156 |
| C4- C5 | 1.378(5) | 1.4179 | 1.4174 | 1.4196 | 1.4488 |
| C4- C9 | 1.396(5) | 1.3916 | 1.3899 | 1.4016 | 1.3997 |
| C5- C6 | 1.386(5) | 1.3919 | 1.3936 | 1.4016 | 1.3979 |
| C6 -C7 | 1.364(6) | 1.3883 | 1.4097 | 1.4076 | 1.3874 |
| C7 -C8 | 1.412(7) | 1.4103 | 1.3901 | 1.4157 | 1.4044 |
| C8 -C9 | 1.371(6) | 1.4103 | 1.4084 | 1.4076 | 1.3908 |
| Bond Angles (°) | X-ray | DBZ | DBZW | DBZI | DBZIW |
| C1- N1- C5 | 108.1(3) | 109.3977 | 109.8646 | 108.3002 | 108.0977 |
| C1- N1- C2 | 125.8(3) | 123.5164 | 124.1667 | 123.5946 | 126.1173 |
| C5- N1- C2 | 126.1(3) | 127.0859 | 125.0423 | 125.9256 | 125.785 |
| C1- N2- C4 | 108.0(3) | 109.3322 | 109.55 | 108.3028 | 108.0663 |
| C1- N2- C3 | 126.3(3) | 125.4505 | 123.0036 | 123.6115 | 125.9066 |
| C4- N2- C3 | 125.7(3) | 125.2173 | 124.6258 | 125.9131 | 126.0094 |
| N1- C1- N2 | 110.2(3) | 106.7967 | 104.9892 | 109.0936 | 110.5965 |
| C5- C4- N2 | 106.9(3) | 107.4000 | 107.1278 | 106.7765 | 106.5914 |
| C5- C4- C9 | 121.9(3) | 121.4302 | 121.0412 | 121.4453 | 120.5588 |
| N2- C4- C9 | 131.1(3) | 131.1698 | 131.8198 | 131.7777 | 132.8475 |
| C4- C5- C6 | 122.2(3) | 120.7732 | 121.046 | 121.4425 | 120.8197 |
| C4- C5- N1 | 106.7(3) | 107.0734 | 107.298 | 106.7731 | 106.6475 |
| C6- C5- N1 | 131.2(3) | 132.1534 | 131.6411 | 131.7839 | 132.5317 |
| C7- C6- C5 | 116.3(4) | 117.6327 | 117.6012 | 117.1697 | 117.5386 |
| C6- C7- C8 | 121.9(4) | 121.4985 | 121.2589 | 121.3859 | 121.5767 |
| C9-C8- C7 | 121.7(4) | 121.1995 | 121.278 | 121.3861 | 122.4264 |
| C8-C9- C4 | 115.9(4) | 117.4659 | 117.7618 | 117.169 | 117.0779 |
| H11-OW1- H12 | 117(2) | | 105.1749 | | 103.235 |
As of Table 3, The data showed that all of the optimized bond lengths and bond angles were a little bit bigger than the values obtained through experimentation. because the computational theoretical data related to the isolated molecule in the gas phase while the experimental data were obtained in the solid phase. The highest bond length difference is 0.063 Å (DBZ), 0.0785 Å (DBZW), 0.0494 Å (DBZI) and 0.0844 Å for the N2-C1 bond, while the biggest bond angle deviation occurred in the N1-C1-N2 at angle 3.433° in DBZ and 5.248° in DBZW, respectively, while in DBZI, the biggest bond angle deviation occurred in the C1-N2-C3 at angle 2.7185° and at the N2-C4-C9 angle (1.7175°) in DBZIW. The root means square error (RMSE) of the DBZ, DBZW, DBZI, and DBZWI is found to be approximately 0.032, 0.039, 0.029, and 0.039, respectively. This result shows that the theoretically calculated bond lengths (using the B3LYP method) have the strongest correlations with experimental values. For bond angles, DBZ, DBZW, DBZI, and DBZWI, respectively, have root mean square errors of 0.819°, 0.856°, 0.594°, and 0.999°. Figures 9 and 10 show, respectively, the correlation between experimental and theoretical bond lengths and bond angles of the molecules DBZ, DBZW, DBZI, and DBZWI.
3.3.2 Mulliken Charge Distributions
The Mulliken population analysis was used to determine the atomic charge values of the molecules (DBZ, DBZW, DBZI, and DBZWI), which are summarised in Table 4 and visually depicted in Fig. 11. The charge on hydrogen atoms is all positive. It should be noted that the DBZI and DBZWI molecules have an iodine atom and, in comparison to the other atoms in the molecule, have the highest negative charges of -0.60193 and − 0.74247, respectively.
The water molecules are backbone in the DBZW and DBZWI in the molecule and because of this has both DBZW (-0.53454) and DBZIW (-0.46966) oxygen atoms were negatively charged. In all benzimidazole derivatives, all nitrogen atoms had a negative charge whereas all hydrogen atoms had a positive charge. Donor and acceptor atoms are signs that intra- and intermolecular hydrogen bonds are present in the solid-state phase.
Table 4
Mulliken charges (e) for the atoms of DBZ, DBZW, DBZI and DBZWI molecules
| ATOMS | DBZ | DBZW | DBZI | DBZIW |
| I | | | -0.60193 | -0.74247 |
| N1 | -0.03461 | -0.002774 | -0.1289 | -0.09337 |
| N2 | -0.01864 | -0.02806 | -0.12916 | -0.104 |
| C1 | -0.07311 | -0.03283 | -0.19938 | 0.028577 |
| H1 | 0.128545 | 0.116349 | 0.338816 | 0.226445 |
| C2 | -0.2554 | -0.26967 | -0.56808 | -0.09805 |
| H21 | 0.103443 | 0.194071 | 0.279819 | 0.159468 |
| H22 | 0.115347 | 0.15801 | 0.21308 | 0.109528 |
| H23 | 0.115182 | 0.143092 | 0.225498 | 0.105733 |
| C3 | -0.39278 | -0.25423 | -0.56796 | -0.08762 |
| H31 | 0.095871 | 0.142793 | 0.27992 | 0.126884 |
| H32 | 0.149501 | 0.146388 | 0.225396 | 0.106709 |
| H33 | 0.16318 | 0.164884 | 0.213102 | 0.113662 |
| C4 | 0.116478 | 0.405221 | 0.276394 | -0.06205 |
| C5 | 0.368051 | -0.02368 | 0.27641 | -0.03217 |
| C6 | -0.2864 | -0.44445 | -0.32647 | -0.10556 |
| H6 | 0.163157 | 0.139092 | 0.235951 | 0.194352 |
| C7 | -0.406 | -0.35792 | -0.20369 | -0.17555 |
| H7 | 0.155808 | 0.123774 | 0.227746 | 0.273841 |
| C8 | -0.26451 | -0.33676 | -0.20365 | -0.08115 |
| H8 | 0.162911 | 0.120627 | 0.227727 | 0.157892 |
| C9 | -0.2689 | -0.26207 | -0.32656 | -0.1275 |
| H9 | 0.162882 | 0.108327 | 0.235901 | 0.139896 |
| OW1 | | -0.53454 | | -0.46966 |
| H11 | | 0.252062 | | 0.188588 |
| H12 | | 0.326734 | | 0.247576 |
FMO analysis
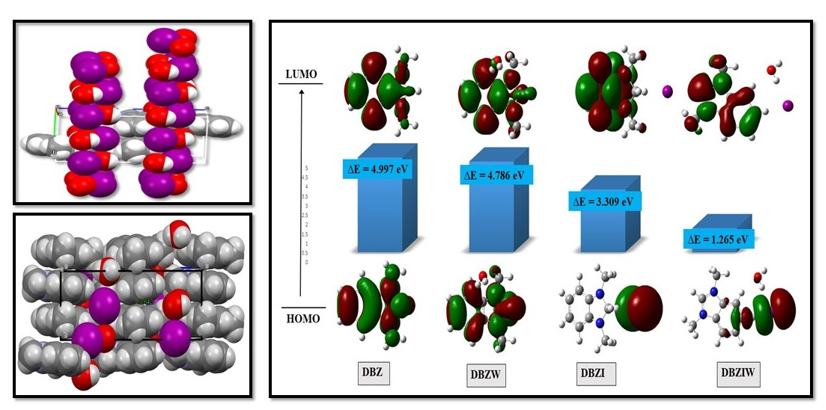
In order to comprehend the frontier effect of water and iodine, if present in benzimidazole derivatives, the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) studies of the benzimidazole derivatives, (a) DBZ, (b) DBZW, (c) DBZI, and (d) DBZIW were conducted. HOMO-LUMO explains a variety of reactions in conjugated systems and is used to predict the most reactive location in π-electron systems using the frontier electron density. FMO analysis is commonly used to describe the optical and electrical properties of organic compounds. To understand the nature of the electronic transition, the electron density plots of HOMO and LUMO are presented in Fig. 8. The energy band gap value of HOMO and LUMO of DBZ, DBZW, DBZI and DBZIW have 4.997 eV, 4.786 eV, 3.309 eV and 1.265 eV, respectively. In molecular interactions, the HOMO stands for electron donors, and its energy is related to the ionization potential (IP), while the LUMO stands for electron acceptors, and its energy is related to the electron affinity (EA). The HOMO-LUMO energy gap, which is useful in determining the molecular electrical transport properties, explains the charge transfer interaction inside the molecule [32–34]. Table 5 lists the predicted HOMO and LUMO energies as well as additional characteristics. Thus, it is clear from Table 5 that DBZ is hard, more stable and less reactive, while compound BDZIW is soft and the least stable and more reactive compare to others. The HOMO-LUMO energy gap decreases from molecule DBZ to DBZW to DBZI and the minimum energy gap is achieved with Water and I substituent in DBZ. Thus, this substituent increases the reactivity of the benzimidazole derivative.
Table 5
HOMO and LUMO energies and global reactivity descriptors calculated for DBZ, DBZW, DBZI and DBZIW
| Parameters | DBZ | DBZW | DBZI | DBZWI |
| HOMO (eV) | -4.800 | -5.232 | -4.946 | -4.006 |
| LUMO (eV) | 0.197 | -0.445 | -1.637 | -2.740 |
| Energy Gap (eV) | 4.997 | 4.786 | 3.309 | 1.265 |
| Ionization Potential | 4.800 | 5.232 | 4.946 | 4.006 |
| Electron affinity | -0.197 | 0.445 | 1.637 | 2.740 |
| Chemical Potential µ | 2.301 | 2.838 | 3.291 | 3.373 |
| Electron negativity χ (eV) | -2.301 | -2.838 | -3.291 | -3.373 |
| Hardness η (eV) | 2.499 | 2.393 | 1.655 | 0.633 |
| Softness (S) | 0.400 | 0.418 | 0.604 | 1.581 |
| Electrophilicity index (ω) | 6.617 | 9.641 | 8.962 | 3.599 |
| Maximum electronic charge | -0.921 | -1.186 | -1.989 | -5.332 |
| Total Energy (A.U.) | -459.784 | -536.255 | -470.413 | 0.116 |
| Dipole moment (Debye) | 2.303 | 4.777 | 9.037 | 21.894 |
{kind=link}