General methods and instruments
Reagents and solvents were purchased from Sigma Aldrich LTDA, Brazil, and used without further purification. Reactions were monitored by thin-layer chromatography (TLC) using silica gel 60 UV-254 pre-coated silica gel plates from MERCK®. Detection was by means of a UV lamp. Flash column chromatography was performed on 300–400 mesh silica gel. Nuclear Magnetic Resonance (NMR), one-dimensional and two-dimensional analyzes of 1H and 13C nuclei were acquired with Bruker Avance spectrometers (USA) model DPX-300 MHz and DRX-600 MHZ and Bruker Ultra-Shield 300 spectrometer FT-NMR. Chemical shifts are given in ppm referenced to internal standards (s = singlet, singlet, d = doublet, t = triplet, m = multiplet); the coupling constant (J), described in Hertz (Hz); and the number of hydrogens deduced from the relative integral.
Infrared (IR) spectra were obtained on a Fourier Transform Infrared Spectrometer (FTIR) Fourier Transform Infrared Spectrophotometer, IRTracer-100 (Shimadzu), in the spectral region from 4000 to 500 cm− 1. High resolution mass spectroscopy (HRMS) data were obtained on a Bruker Daltonics mass spectrometer, model microOTOF-Q II - ESI-Qq-TOF and 4000 QTRAP AB SCIEX. Melting points were determined on a Buchi M-560 melting point apparatus BUCHI Brazil Ltda
Measurement of [18F] radioactivity was performed using a Carpintec dose calibrator (CRC-15R, New Jersey, USA). The Agilent High Performance Liquid Chromatography (HPLC) system (USA) was used for quality control of compounds labeled with [18F]. The system is equipped with a model 1260 quaternary pump, a model 1260 UV absorbance detector and a radioactivity detector (Raytest, Germany). Agilent Chem Station software was used to operate Agilent HPLC systems. The HPLC equipment with UV and radioactivity detector, as well as the column used for analysis (ZORBAX Eclipse Plus C18 Analytical 4.6 x 250 mm, 5 µm) are from Agilent Technologies, Brazil.
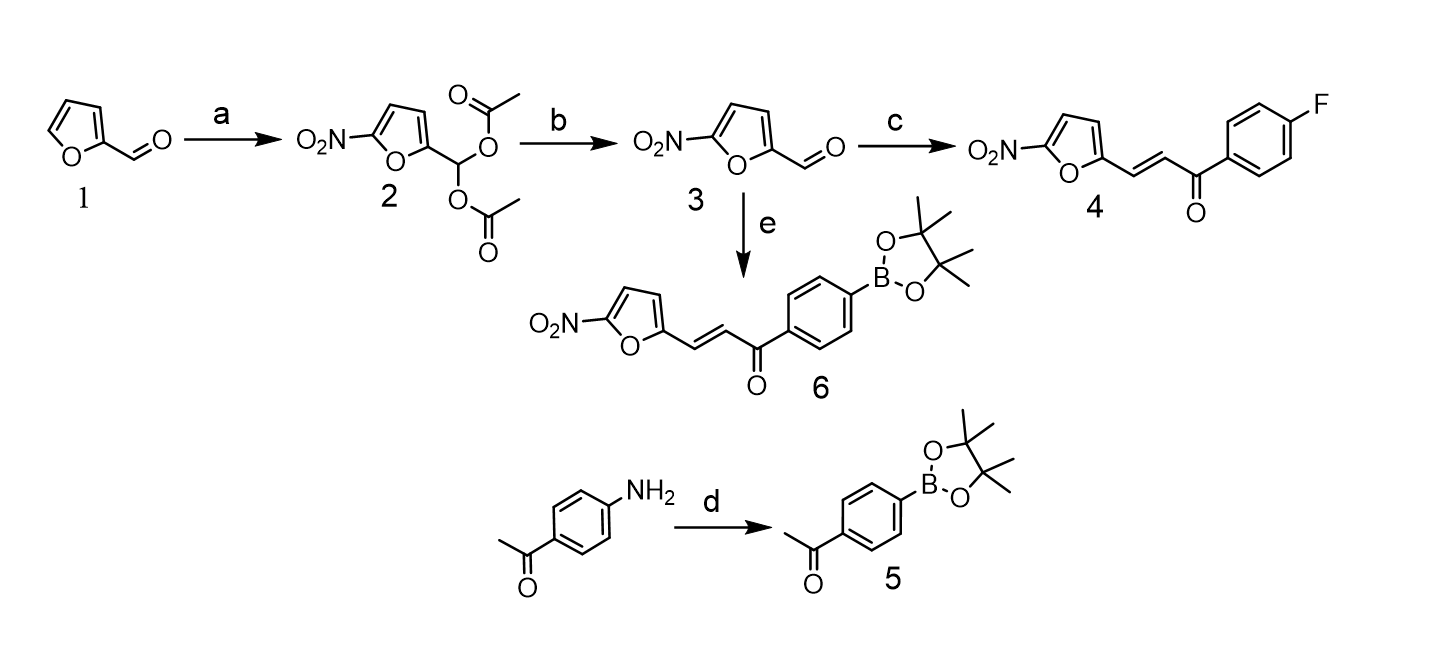
Chemical synthesis
Synthesis of 5-nitro-2-furaldehyde diacetate
Furfural was nitrated following the procedure reported by Taimoory, S. Maryamdokht et al. with some modifications (34). Briefly, Acetic anhydride (180 mL) was placed in a dried 500-mL three-necked, round-bottomed flask equipped with a mechanical stirrer and a thermometer, and afterwards the mixture was then cooled to -10ºC in a benzyl alcohol/dry ice bath. 17.2 mL of concentrated HNO3 and 0.15 mL of concentrated H2SO4 were mixed in a small beaker, and this solution was slowly and carefully added into the chilled acetic anhydride while stirring between − 10ºC and − 5ºC. Afterwards, 20.8 mL (0.251 mol) of freshly distilled furfural was added dropwise over 20 minutes into the cold acid mixture with stirring at the same temperature, and the resulting solution was left to stir for 1 h between − 10ºC and − 5ºC. After this time 160 mL of water were added, and the mixture was left to stir at room temperature for 30 min, over which a white precipitate formed. A 10% NaOH solution was added to the mixture until the pH rose to 2.5–2.7, and it was warmed on a water bath at 55ºC for 1h. The mixture was poured into crushed ice, stirred for 10 min and the white precipitate formed filtered, washed with water, recrystallized twice from ethanol, and air-dried to provide 36.63 g of the product as a white crystalline solid in 60% yield. 1H NMR (300 MHz, Chloroform-d) δ 7.73 (s, 1H), 7.30 (d, J = 3.6 Hz, 1H), 6.75 (d, J = 3.6 Hz, 1H), 2.19 (s, 6H) (Supplementary file 1). m.p. 90–91 ºC.
Synthesis of 5-nitro-2-furaldehyde
5-nitro-2-furaldehyde was obtained following a procedure reported by Mohammadpoor-Baltork, I. and Aliyan, H. (35). In short, to a solution of 5-Nitro-2-furaldehyde diacetate (4.864 g, 20 mmol) in acetonitrile (100 mL), was added SnCI2.2H20 (902.52 mg, 4 mmol, 20% mol) and the clear mixture was refluxed for 1h (turned dark after 20 min of reaction) when TLC (100% toluene) indicated completion. The solvent was evaporated, and the residue was purified by flash column chromatography eluting with chloroform to give 2.54 g of 5-nitro-2- furaldehyde as a yellow solid in 90% yield. 1H-NMR (600 MHz, Chloroform-d) δ 9.85 (s, 1H), 7.44 (dd, J = 3.8, 0.4 Hz, 1H), 7.38 (d, J = 3.8 Hz, 1H) (Supplementary file 2). m.p. 38-38.7ºC.
Synthesis of (E)-1-(4-fluorophenyl)-3-(5-nitrofuran-2-yl)prop-2-en-1-one, [19F]-FNFP
Following the methodology reported by Tawari, Nilesh R et al. (24), 5-nitro-2-furaldehyde (988 mg, 7 mmol) was dissolved in 10 mL of glacial acetic acid at room temperature. 400 µL of concentrated sulfuric acid and 4-fluoroacetophenone (7 mmol, 846 µL) were added in sequence, and the mixture was stirred at room temperature overnight. Ice-cold water (50 mL) was added, the solid was recovered by filtration and washed carefully with ice cold ethanol. The product was then recrystallized from ethanol to yield 1.37 g of (E)-1-(4-fluorophenyl)-3-(5-nitrofuran- 2-yl)prop-2-en-1-one as yellow needles in 75% yield. 1H-NMR (600 MHz, DMSO-d6) δ 8.26–8.20 (m, 2H), 7.89 (d, J = 15.6 Hz, 1H), 7.83 (d, J = 3.9 Hz, 1H), 7.61 (d, J = 15.6 Hz, 1H), 7.47 (d, J = 3.9 Hz, 1H), 7.45–7.41 (m, 2H) (Supplementary file 3). m.p. 183–184ºC.
Synthesis of 4-acetyl phenylboronic acid pinacol ester
Following the procedure reported by Mo, Fanyang et al. (36), 4-aminobenzophenone (1.352 g, 10 mmol), bis(pinacolato)diboron (2.667 g, 10.5 mmol, 1.05 equiv.) and benzoyl peroxide (20.4 mg, 0.08426 mmol, 2% mol) were weighted in a 50 mL round-bottom flask. To this mixture, anhydrous acetonitrile (15 mL) was added, and then tert-butyl nitrite (1.785 mL, 15 mmol, 1.5 equiv.) in 5 mL of anhydrous acetonitrile was added dropwise at room temperature with stirring. The resulting mixture was stirred at room temperature overnight, and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography eluting with hexanes/ethyl acetate 1/50 to 1/20 to give 1.6 g of 4-acetyl phenylboronic acid pinacol ester as a white-off crystalline solid in 65% yield. 1H-NMR (600 MHz, Chloroform-d) δ 7.85–7.77 (m, 4H), 2.51 (s, 3H), 1.26 (s, 12H) (Supplementary file 4). m.p. 59–60ºC.
Synthesis of (E)-(4-(3-(5-nitrofuran-2-yl)acryloyl)phenyl)boronic acid pinacol ester: 5-nitro-2-furaldehyde (168 mg, 1.2 mmol) was dissolved in 1.5 mL of glacial acetic acid at room temperature. 120 µL of concentrated sulfuric acid and 4-acetyl phenylboronic acid pinacol ester (295.3 mg, 1.2 mmol) were added in sequence, and the mixture was stirred at room temperature overnight. Ice-cold water (5 mL) was added, the solid was recovered by filtration and washed carefully with ice cold ethanol. The product was then recrystallized from ethanol to yield 319 mg of (E)-(4-(3-(5- nitrofuran-2-yl)acryloyl)phenyl)boronic acid pinacol as yellow needles in 72% yield. 1H-NMR (600 MHz, DMSO-d6) δ 8.12–8.10 (m, 2H), 7.89–7.84 (m, 3H), 7.83 (d, J = 3.9 Hz, 1H), 7.62 (d, J = 15.6 Hz, 1H), 7.47 (d, J = 3.9 Hz, 1H), 1.33 (s, 12H) (Supplementary file 5). 13C-NMR DEPTQ (151 MHz, DMSO) δ 153.60, 152.50, 139.30, 135.30, 129.41, 128.28, 125.31, 118.52, 115.26, 84.62, 25.15 (Supplementary file 6). m.p. 170–171ºC. ESI-mass spectrometry. [M + Na]+=392,1274 expected MW = 369,18 g/mol (Supplementary file 7). Infrared spectra: ʋ (= C-H) aromatic 3118 cm− 1; ʋ (C-H) 2929 cm− 1; carbonyl stretching vibration for the enone (= C-C = O) 1661 cm− 1; N–O asymmetric stretch 1479 cm− 1, N–O symmetric stretch 1365 cm− 1 (Supplementary file 8).
Radiochemistry
No-carrier-added [18F]F− was produced by 18O(p,n)18F reaction in an 18-MeV cyclotron (IBA, Belgium). Sep-Pak Light QMA cartridges (Waters, Brazil) were pre-conditioned using two different methods: 1) 10 mL of 0.5 M K2CO3 solution, followed by 20 mL of Milli-Q water and purged with 5 mL of air; 2) 10 mL of 0.5M potassium triflate (KOTf), followed by 20 mL of Milli-Q water and purged with 5 mL of air.
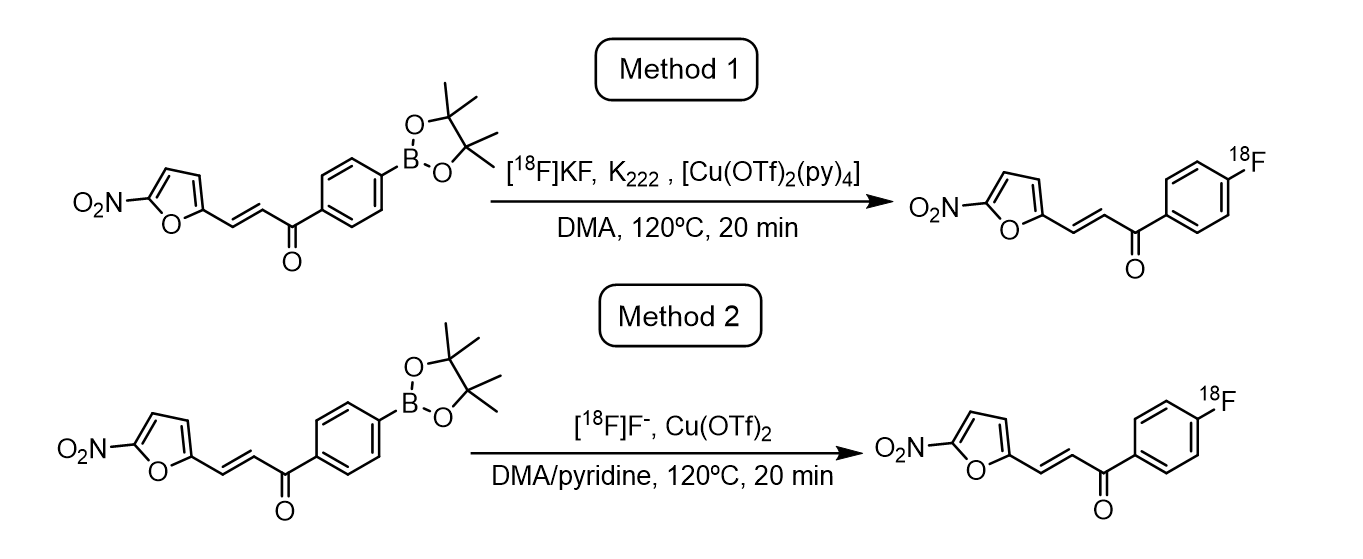
Radiosynthesis of (E)-1-(4-[18F]fluorophenyl)-3-(5-nitrofuran-2-yl)prop-2-en-1-one [18F]FNFP
Two different methodologies were studied to prepare [18F]FNFP manually.
Method 1, following the procedure reported by Tredwell, Matthew et al. (25). In short, the fluoride ion [18F]− solution was trapped in an anionic Sep Pak light QMA cartridge, previously conditioned with K2CO3 0.5 M and water, and the [18F]− was then eluted into the reaction vial with a mixture of K2CO3 (2.8 mg; 20 µmol) and Kryptofix 2.2.2 (15 mg; 40 µmol) in 1 mL of acetonitrile/water (8:2). Then, the solution was subjected to three cycles of azeotropic distillation with anhydrous acetonitrile (1mL) at 100°C under a nitrogen flow. Afterwards, (E)-(4-(3-(5-nitrofuran-2-yl)acryloyl)phenyl)boronic acid pinacol ester (4.8 mg, 13 µmol) in 150 µL of anhydrous DMA and [Cu(OTf)2(py)4] (8.8 mg, 13 µmol) in 150 µL of DMA were added and the resulting solution was stirred at 120ºC for 20 min under air, quenched with water (300 µL) and an aliquot was removed for analysis by radio-HPLC to calculate the RCY and identify the product, respectively.
Method 2, according to a procedure reported by Lahdenpohja, S.O., Rajala, N.A., Rajander, J. et al. (26). Succinctly, the fluoride ion [18F]− solution (80–100 mCi, approximately 1–2 mL) was diluted in 8–10 mL of fresh miliQ water in a flask. This solution was taken up in a 20 mL syringe and passed through a pre-conditioned anionic QMA cartridge, previously conditioned with KOTf 0.5 M and water. The cartridge was slowly washed with 5 mL of anhydrous DMA, and the [18F]− was then eluted with a solution of Cu(OTf)2 (34.7 mg, 96 µmol) in anhydrous DMA (0.5 mL) into the reaction vial already containing (E)-(4-(3-(5-nitrofuran-2-yl)acryloyl)phenyl)boronic acid pinacol ester (5.5 mg, 15 µmol) diluted in 50 µL of anhydrous DMA and 50 µL of anhydrous pyridine. The QMA cartridge was washed with 0.5 mL of anhydrous DMA into the reaction vial, giving a total volume of 1.1 mL, and the resulting blue solution was stirred at 120ºC for 20 min under air. The reaction was quenched with water (500 µL) and an aliquot was removed for analysis by radio-HPLC to calculate the RCY and identify the product, respectively.
After HPLC identification of the product, [18F]FNFP crude reaction mixture was then taken up in water (20 mL) and passed through a pre-conditioned C18 plus cartridge. The product was then eluted from the C18 with 1 mL of ethanol and purified by semipreparative HPLC using MiliQ water (0.1% trifluoroacetic acid), channel A, and acetonitrile, channel B, with a gradient run using 10%-100% of B in 20 minutes, with a flux of 3 mL/min, retention time 14–15 min.
{kind=link}
{kind=link}