We developed the MOSBY model that learned a mapping from RetCCL (contrastive self-supervised learning)-based WSI features to bulk transcriptome, proteome, whole exome, SNP-array, and DNA methylome based profiles (Fig. 1a). We limited our study to 21 TCGA indications14 with at least 200 paired RNA-seq and H&E whole slide images, resulting in a total of 12592, 10192, and 12090 images matching with transcriptome, proteome and DNA-based data respectively (breakdown by indication in Supplementary Table 1). Analyzed transcriptomic features consisted of 55 TME-related genes15,16 (Supplementary Table 2) and 175 gene sets that covered tumor-related processes17–32, metabolic pathways33, and TME cell type or process signatures34–37 (Supplementary Table 3). The proteome model involved 191 total and phosphoprotein antibodies from the TCGA reverse-phase protein array (RPPA) panel that focused on tumor-intrinsic and oncogenic processes 38. Tested DNA-based features were limited to tumor purity, cancer DNA fraction, subclonal genome fraction, and average DNA methylation (all continuous measures bound to the interval [0,1]).
In addition to single indication models, we also trained MOSBY in pan-tissue, pan-cancer, pan-squamous and pan-adenocarcinoma settings. Pan-tissue models consisted of LUNG (lung adenocarcinoma and squamous cell carcinoma), KIDNEY (clear cell and papillary renal cell carcinoma), BRAIN (glioblastoma and low grade glioma), and COADREAD (colon and rectal adenocarcinoma). The pan-adenocarcinoma (ADENO) model consisted of pancreatic, lung, stomach, colon, rectal, prostate, and ovarian adenocarcinomas; while the pan-SQUAMOUS model included squamous cell carcinomas of the lung, cervix, and head and neck. The ADENO and SQUAMOUS models allowed us to investigate whether histological similarities beyond tissue architecture enabled MOSBY to learn a better mapping from WSI features to multi-omic data.
Similar to the HE2RNA12 model, a 2-layer perceptron was adopted as the multi-output regression network that mapped image features to omic variables, and a maximum of 8000 image tiles per slide were used. Separate models were trained for gene, signature, protein, and DNA variables. In contrast to HE2RNA, image tiles were randomly selected from each WSI to capture an unbiased representation of the entire slide, and were all used in training without clustering. In addition, batch-normalized transcriptomic and proteomic data were used as ground truth to enable across-indication comparisons with resulting MOSBY predictions. To obtain slide-level predictions, tile-level predictions of omic features were aggregated by averaging.
Contrastive self-supervised pretraining benefits prediction of omic data from H&E whole slide images
The RetCCL feature extractor utilized TCGA H&E images during contrastive training, however it was not supervised by gene expression information. Hence, there are no a priori guarantees for RetCCL-based image features to predict gene expression with higher accuracy than ImageNet-based features. The success in this task hinges upon the diversity of semantic (in this case, biological) features in the representations learned by RetCCL. Thus we investigated the performance differences between MOSBY models trained with RetCCL- or ImageNet-based image features. Models were trained with 5-fold cross-validation (80/20 percent training/test set split). A random one fifth of the training set was also allocated as validation set to determine an early stopping criterion using the Spearman correlation between slide-level model prediction and ground truth omic data. Spearman correlation was preferred to mitigate the effects of outlier samples in relatively smaller cohorts. Correlation coefficients from test sets were subsequently averaged across five folds to obtain the final performance score for a given feature.
RetCCL-based image features consistently led to higher cross-validated test set averages in all four omic data types compared to features extracted with ImageNet-pretrained ResNet-50 architecture (Fig. 1b-1e). The pan-cancer model (PANCAN) with RetCCL-based features achieved the highest performance for all tested data types with median cross-validated Spearman correlation of 0.722 in single genes (0.673 with ImageNet-features) (Fig. 1b), 0.693 in signatures (0.647 with ImageNet-features) (Fig. 1c), 0.549 in proteomic data (0.512 with ImageNet-features) (Fig. 1d), and 0.6 in DNA features (0.533 with ImageNet-features) (Fig. 1e). In terms of single indication models, the thyroid cancer model (THCA) achieved best performance for single gene and protein expression data sets with RetCCL-based features. Median cross-validated Spearman correlations in these models were 0.59 vs 0.524 for single gene, and 0.443 vs 0.385 for protein expression data with RetCCL and ImageNet-based features respectively (Fig. 1b, 1d). The single indication models achieving best performance for signature and DNA data were the liver and bladder cancer models respectively (LIHC and BLCA) (Fig. 1c, 1e). Across tested signatures, the LIHC model showed a median cross-validated Spearman correlation of 0.532 with RetCCL-based vs 0.45 with ImageNet-based features. For tested DNA features, the BLCA model achieved a median cross-validated Spearman correlation of 0.553 with RetCCL-based vs 0.417 with ImageNet-based features. Taken together, these results indicated that contrastive self-supervised pretraining in large-scale histology datasets benefits prediction of multiomic data from H&E-stained whole slide images.
MOSBY tile level predictions are validated with spatially resolved transcriptomic data
For inference tasks, MOSBY ‘full models’ were trained with data from 80% of patients, with the remaining 20% used as the validation set to determine an early stopping criterion. An MOSBY full model was trained in the TCGA breast cancer (BRCA) cohort (N = 1576 WSIs), and subsequently deployed on H&E image tiles from a publicly available spatially resolved transcriptomic breast cancer dataset39 (referred to as ST from here on, N = 68 WSIs) that served as an independent validation cohort. Image tiles were centered around ST spot coordinates to enable one-to-one comparison between spot level ground truth data and tile level model predictions (Methods). For individual slides, the MOSBY signature model predictions showed the highest concordance with ground truth for ‘poor differentiation’ (Stemness_Kim_Myc, Spearman r = 0.71) and stromal features (Stroma_Estimate, Spearman r = 0.63) (Fig. 1f, Supplementary Fig. 1a). Across 68 slides, concordance was highest for a monocyte signature with a median Spearman correlation of 0.238 (Supplementary Fig. 1a), and a maximum of 0.611 (Fig. 1f). This large variation across slides was observed for all tested signatures suggesting that the quality of spatially resolved data is critical in validating MOSBY predictions. Of note, CD8 T cell infiltration and proliferation-related model predictions also showed strong concordance with ground truth, demonstrating the variety in phenotypes captured successfully by the model (Supplementary Fig. 1b).
Spot level concordance for single gene expression values was overall lower than that for signatures (Supplementary Fig. 1d), potentially driven by the large degree of zero reads (i.e. dropouts) in ground truth data (Supplementary Fig. 1e). However, model predictions for genes associated with stroma, plasma cells, and epithelial features showed good performance for individual slides (COL1A2, MZB1, EPCAM respectively) (Fig. 1g). CD68, a myeloid marker, showed the highest median concordance in gene models (Spearman r = 0.127), which was low in magnitude but consistent with the high concordance of myeloid cells in signature models (Supplementary Fig. 1d).
As MOSBY aims to discover the joint latent space between H&E WSIs and bulk omic profiles, the correlation structure in ground truth data was also utilized to validate MOSBY model predictions (Methods). This analysis revealed that the MOSBY signature-signature correlation matrix was significantly different from random for 67 out of 68 slides (empirical p-value < 0.05) (Supplementary Fig. 1c). For gene-gene correlation matrices, this value dropped to only 28 out of 68 slides (Supplementary Fig. 1f), again highlighting that computing gene signature scores is an effective strategy to deal with the dropout issue in spatially resolved transcriptomic datasets. Despite reduced overall performance in gene models, predicted gene expression levels still successfully captured specific highly correlated gene blocks derived from ground truth data, such as those among cancer-associated fibroblast (CAF) and immune cell marker genes (Supplementary Fig. 1g). An analysis across 68 slides indicated that MOSBY model predictions were particularly good at distinguishing CAF-related features from other TME population marker genes. For 63 out of 68 slides, the MOSBY gene-gene correlation matrix had a cluster significantly enriched for fibroblast marker genes, and this enrichment was higher than 89% for the large majority of the slides (Supplementary Fig. 1h).
Serially sectioned CD8 IHC whole slide images validate MOSBY in silico spatialization
MOSBY tile-level predictions were further validated with CD8 antibody-stained immunohistochemistry (IHC) WSIs. To this end, gene and signature models were trained with data from urothelial carcinoma immune checkpoint inhibitor (ICI) trials IMvigor21040 and IMvigor21141 (Methods, N = 1460 paired H&E and RNA-seq samples). H&E-CD8 double-stained slides were not available, thus serially-sectioned single-stained H&E and CD8 IHC whole slide images were computationally aligned (Methods, N = 1017 paired H&E and CD8 IHC WSIs). 454 slides that were correctly aligned based on multiple alignment metrics were subjected to a manual QC check and a stringent 95% tissue overlap threshold (Methods, Fig. 2a). The remaining 42 IHC WSIs constituted the analysis set, and were used to categorize “brownness” of pixels based on diaminobenzidine staining (i.e. DAB mask)(Methods). Each DAB mask was split into tiles, and “brown” pixels in each tile were counted to determine tile-level CD8 IHC quantification (Methods). Pearson correlation coefficient was then computed between CD8 IHC and MOSBY-predicted tile-level values (Fig. 2a).
To compare with CD8 IHC quantification, gene features CD8A, CD8B, and signature features Cibersort_CD8_T_cells and T_effector_cells were used from MOSBY model predictions. Across 42 slides, CD8B and Cibersort_CD8_T_cells showed the strongest concordance with CD8 IHC, having the highest correlation in 22 and 15 slides respectively (Fig. 2b). Overall, CD8B model predictions had the highest median correlation with CD8 IHC tile-level values (Pearson r = 0.352), with a range of [-0.006, 0.789]. The density plot for r = 0.789 correlation (N = 16859 tiles, p ≈ 0, two-tailed exact test) indicates stronger concordance for values closer to the higher and lower ends of the distribution (Fig. 2c). Visual inspection of CD8 IHC and H&E WSIs confirmed successful alignment, but also revealed minor differences between tissue sections potentially due to these being non-adjacent sections (Fig. 2d). At low magnification, visualization of DAB mask tile level quantification and CD8B model predictions indicated that the model successfully captures CD8 T cell infiltration patterns in the tumor (Fig. 2e, 2f). High magnification inspection of model predictions and DAB mask (as well as the input H&E and CD8 IHC images) confirmed that MOSBY, despite being weakly supervised on a patient level bulk RNA-seq dataset, effectively learned a CD8 T cell-specific representation in H&E images (Fig. 2g–2j).
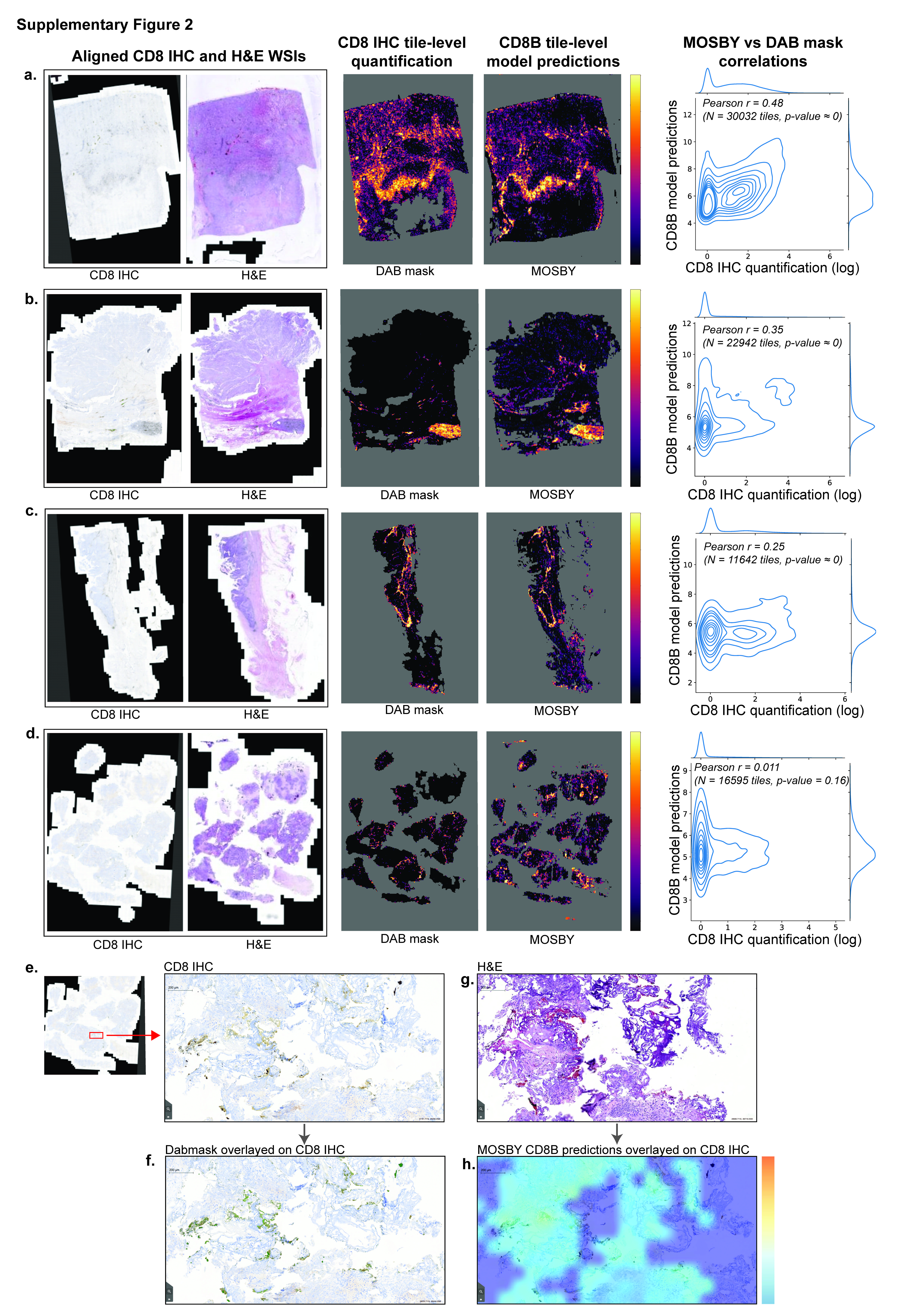
In order to gain insight into both high and low concordance cases, four representative slides were selected based on the distribution of predicted CD8B vs DAB mask correlations (near the minimum, and 25th, 50th, and 75th percentiles). Selected slides had Pearson correlation values 0.48 (N = 30032 tiles, p ≈ 0, two-tailed exact test), 0.35 (N = 22942 tiles, p ≈ 0, two-tailed exact test), 0.25 N = 11642 tiles, p ≈ 0, two-tailed exact test, and 0.011 (N = 16595 tiles, p = 0.16, two-tailed exact test) respectively (Supplementary Fig. 2a–2d). Low magnification inspection of DAB mask and MOSBY CD8B predictions indicated that model predictions captured overall CD8 T cell infiltration patterns in the first three slides despite the correlation being as low as 0.25 (Supplementary Fig. 2a–2c). For the last slide (r = 0.011), spatial patterns in DAB mask and model predictions were conspicuously discordant (Supplementary Fig. 2d). High magnification inspection of CD8 IHC and DAB mask images from this last slide revealed that DAB mask captured brown staining artifacts in regions that did not show any expected CD8-specific staining patterns (Supplementary Fig. 2e, 2f). In addition, CD8 IHC and H&E tissue sections showed misalignment in small substructures, despite an overall high score in tissue alignment (Supplementary Fig. 2e-2h). Taken together, these results showed that MOSBY successfully learned a CD8 T cell-specific representation on H&E images, and correlations as low as 0.25 may be sufficient to capture overall CD8 T cell infiltration patterns in the tumor microenvironment, suggesting the potential of this tool for patient stratification in ICI trials.
Validation with pathologist annotations of cancer regions indicates epithelial gene and signature markers may be misleading in accounting for tumor-specific expression
Various RNA- and protein-based epithelial markers such as EPCAM (GENE) and E-cadherin (PROTEIN) are frequently used in the literature to mark cancer cells in tissue specimens. However, epithelial markers can be expressed in both transformed and non-transformed epithelial cells. We utilized MOSBY tile level predictions in order to compare virtual spatialization of epithelial markers with pathologist tumor region annotations and assess the tumor-specificity of commonly used tumor markers.
MOSBY full models were trained in TCGA lung adenocarcinoma (LUAD), and deployed on WSIs in IMpower15043, an independent validation cohort in nonsquamous nonsmall cell lung cancer with pathologist annotations (i.e. pen marks) demarcating cancer epithelia.
Pathologist annotations were compared with MOSBY features of tumor purity (DNA-based) and epithelial markers EPCAM (GENE), E-cadherin (PROTEIN), and an epithelial gene signature by Taube et al20. WSI level comparison indicated that MOSBY models trained with DNA-based quantification of cancer cells (tumor purity) achieves a stronger concordance with pathologist tumor region annotations (Fig. 3a, 3b). E-cadherin closely followed DNA-based features in terms of concordance with pathologist annotations, albeit noticeably lower specificity for the cancer region in Slide 2 (Fig. 3d vs 3b). Interestingly, predicted spatialization of EPCAM transcript levels showed relatively poor concordance with cancer epithelium annotation (Fig. 3c); and the performance of the Taube et al. epithelial signature was further reduced with highest predicted tiles corresponding to non-tumor regions (Fig. 3e). Collectively, these results suggest that employing DNA-based tumor cellularity estimates is the most reliable approach for accounting for tumor-specific expression in bulk datasets; and published epithelial signatures may not be fit-for-purpose in this task.
MOSBY enables investigation of intratumor heterogeneity with prediction of single gene, signature, and protein expression levels
The MOSBY framework employs RNA-seq and RPPA data to predict gene (or signature) and protein expression levels respectively (Methods). The utility of the model in predicting single gene, signature, and protein expression levels and inferring intratumor heterogeneity is demonstrated in Impower150 in the context of cell proliferation features (Supplementary Fig. 3). Three representative H&E slides are shown in this figure together with pathologist annotations of cancer epithelia (Supplementary Fig. 3a), and MOSBY-predicted spatialization of cell proliferation markers Cyclin B1 (Supplementary Fig. 3b), Hallmark G2M checkpoint signature (Supplementary Fig. 3c) and MKI67 (Supplementary Fig. 3d). Even though tile-level predictions of all three cell cycle markers are highest in cancer epithelia-annotated regions, the spatialization of protein and signature markers (Cyclin B1 and Hallmark G2M checkpoint signature) demonstrate a stronger localization to cancer epithelia (Supplementary Fig. 3b, 3c, 3d). In addition, the spatialization of MKI67 expression in all three slides appears to suffer from checkerboard-like artifacts (Supplementary Fig. 3d). These results suggest that molecular data prediction from WSIs may be more successful with proteins or gene signatures, as opposed to single genes. Potential reasons for this observation are 1) proteins being closer to phenotype than genes and 2) gene signatures increasing the signal-to-noise ratio from RNA-seq data. Biologically, the predicted spatial pattern of Cyclin B1 and Hallmark G2M checkpoint signature levels show differential proliferation levels across cancer epithelia tiles (Slide 3 in Supplementary Fig. 3b, 3c), indicating MOSBY also has potential to infer intratumor heterogeneity.
MOSBY predicts stroma, immune, and proliferation features with highest accuracy in TCGA
Cross-validated test set performance in single-indication signature and protein models was used to determine biological features with the highest prediction accuracy, and those with 25 highest performance are highlighted in Fig. 4a, 4d. The best-performing signatures ESTIMATE Stroma and ESTIMATE Immune had median correlations 0.627 and 0.616 across all tested indications (Fig. 4a). In randomly split test sets, the Spearman correlation of ESTIMATE Stroma and ESTIMATE Immune signatures reached as high as 0.787 and 0.781 in skin cutaneous melanoma and thyroid cancer respectively (Fig. 4b). Other best-performing signatures were again largely enriched in stroma and immune features as well as general mesenchymal characteristics (such as Hallmark EMT and Taube et al mesenchymal signatures). (Fig. 4a). Of note, particular signatures known to play an important role in specific cancer indications also showed strong prediction accuracy in those pertinent settings. For instance, the Hallmark Angiogenesis signature had Spearman r = 0.746 in the liver cancer model (LIHC), and a fatty acid elongation signature had Spearman r = 0.741 in the low grade glioma setting (LGG) (Fig. 4b). After adjusting for multiple hypothesis testing with false discovery rate (adj. p < 0.05), 14 out of 21 tested indications showed significant performance (as measured by Spearman r) for more than 90% of tested signatures (total N = 175 signatures) (Fig. 4c).
In contrast with our signature set that represented both tumor-intrinsic pathways and cell populations in the TME, the TCGA RPPA antibody set was heavily focused on tumor-intrinsic pathways. Therefore, the best-performing proteins had a diversity of representation from tumor-intrinsic characteristics, such as proliferation (Cyclin-B1, FOXM1), DNA repair (MSH6, PARP1), and apoptosis (cleaved Caspase7) (Fig. 4d). MSH6 and Cyclin B1 were the 2 best-performing proteins that were tested in at least 20 indications (Fig. 4d). These proteins had median Spearman r = 0.423 and 0.417 respectively across tested indications, but correlations in individual indications were as high 0.676 in PRAD for Cyclin B1, and 0.593 in LGG for MSH6. Overall, lower prediction accuracy in protein models was expected due to the lower signal-to-noise ratio in the RPPA technology compared to RNA-seq. However, in specific settings such as the PRAD model, both total and phosphoproteins showed strong prediction accuracy in test sets. Progesterone receptor (PR) and cMET_pY1235 reached Spearman r = 0.614 and 0.631 respectively in this indication (Fig. 4e). Also, the AKT/mTOR pathway showed evidence of strong prediction accuracy in the sarcoma setting where S6 and RICTOR_pT1135 antibodies showed Spearman r = 0.731 and 0.695 respectively (Fig. 4e). Despite paucity of representation in the RPPA panel, stromal and immune features were also found among the best-performing proteins such as ECM-associated Collagen-VI, Fibronectin and T cell-associated Lck (Fig. 4d). Overall, 11 out of 20 tested indications showed significant performance (adj. p < 0.1) for more than 90% of tested antibodies (total N = 191 antibodies) (Fig. 4f).
In terms of single gene models, best performing features confirmed the strong prediction accuracy associated with stromal features observed above; the highest-ranking genes were marker genes of fibroblasts (e.g. LUM, COL5A1) (Fig. 4g). Top-ranking genes also included markers of macrophages (e.g. CSF1R), and T cells (e.g. CD3E). In DNA models, tumor cellularity measures (tumor purity, cancer DNA fraction) achieved better performance than subclonal genome fraction, and average DNA methylation features (Fig. 4h).
Multi-indication runs may show exaggerated performance
The results above demonstrated that MOSBY models trained with gene signature and protein expression data generally perform better than those trained with single genes. Thus, using signature and protein expression data, we next addressed whether single- vs multi-indication models (introduced in Fig. 1b) had more utility from a clinical perspective. We focused on performance in the unseen test set to evaluate the generalization capacity of each approach in a scenario emulating the clinical setting; i.e. predicting gene and protein expression levels for a single patient. Performance was measured, as above, with the Spearman correlation between MOSBY slide-level predictions and ground truth levels of signature or (phospho)protein expression (N = 175 for signatures, 191 for total and phosphoproteins).
Multi-indication models (PANCAN, ADENO, BRAIN, KIDNEY, SQUAMOUS, LUNG) showed superior performance for both signature and protein prediction tasks, compared to single-indication counterparts (Supplementary Fig. 4a). The PANCAN model had a median cross-validated Spearman correlation of 0.693 for tested signatures (Supplementary Fig. 4a), with epithelial, proliferation, and stemness signatures showing the best prediction accuracy (cross-validated Spearman r = 0.826, 0.824, and 0.82 respectively). We focused on the Benporath_ES2 stemness signature to demonstrate the bias associated with multi-indication models. In a randomly split test set, this signature had Spearman r = 0.84 between predicted and ground truth levels in the PANCAN dataset (Supplementary Fig. 4b, left). However, the correlations in individual indications varied between 0.244–0.645 for this signature, suggesting that the PANCAN model performance may be spuriously inflated by combining multiple indications with different distributions; reminiscent of the Simpson’s paradox (e.g. OV and READ slides concentrated on the higher end, and THCA and KIRC concentrated on the lower end of the distribution; Spearman r = 0.248, 0.326, 0.41, and 0.482 for OV, READ, THCA and KIRC respectively) (Supplementary Fig. 4b, right).
In cross-validated protein models, Cyclin B1, Claudin-7 and progesterone receptor (PR) showed the best prediction accuracy in the PANCAN model (Spearman r = 0.775, 0.772, and 0.749 respectively). To ask whether this performance was spuriously inflated, we again compared PANCAN and single indication models. In a randomly split test set, Cyclin B1 had Spearman correlation of 0.76 in the PANCAN model (Supplementary Fig. 4c, left), while correlations in individual indications were lower ranging from − 0.10 to 0.597. Simpson’s paradox was again evident in protein model predictions with Cyclin B1 levels in individual indications localizing to smaller parts of the pan-cancer distribution and showing lower prediction accuracy (e.g. Spearman r = − 0.103, 0.198, 0.214, and 0.263 for LIHC, THCA, READ and CESC respectively) (Supplementary Fig. 4c, right).
We then investigated the potential bias in multi-indication models more systematically by comparing median test set performance (i.e. median correlation for signatures or proteins) in single vs multi-indication models (Supplementary Fig. 4e, 4g). The train/validate/test set splits in multi-indication models were implemented with indication stratification, enabling comparison with single-indication training. Results showed that neither single-indication nor multi-indication training was universally better (Supplementary Fig. 4e, 4g). However, evidence also showed that multi-indication training generally did not improve performance if a single-indication model reached a median performance of 0.45 for signatures (Supplementary Fig. 4e), and 0.35 for proteins (Supplementary Fig. 4g), with LGG as a potential exception benefiting from joint training with GBM.
Pan-tissue training appeared to improve median test set accuracy in certain settings such as protein models in kidney, brain, lung, colon/rectum (Supplementary Fig. 4g), and signature models in brain (Supplementary Fig. 4e). However, at the individual signature/protein level, further heterogeneity in model performance was observed (Supplementary Fig. 4f, 4h). A subset of signatures/proteins benefited from multi-indication training whereas another subset did not. This observation prevented us from making general recommendations as to the deployment of single vs multi-indication models in a clinical setting. In circumstances where there is an interest in specific signatures/proteins, it may be suboptimal to make the model decision based on median performance. However, results in Supplementary Fig. 4e, 4g can be used as a guideline in other settings that lack a focus on specific biological features. We utilize single indication models in subsequent parts of this study in order to prevent potential bias from multi-indication training.
Spatial patterns inferred from tile-level MOSBY predictions increase survival predictive power of gene signature-based models
MOSBY tile-level predictions enable in silico spatialization for a tested omic feature, as well as assess spatial correlation between two tested features (positive and negative correlations indicating colocalization and spatial exclusion respectively). We define a ‘colocalization feature’ as the Pearson correlation between tile-level predictions of two omic features. As correlation coefficients were computed across all tiles on a slide, colocalization features represent slide-level as opposed to ‘local’ spatial patterns. We focused on our signature panel (N = 175) as the omic features to derive colocalization features and investigate survival associations, as the signature panel covered both tumor and non-tumor TME components. Moreover, using signatures as opposed to single genes enable the discovery of ‘biologically interpretable’ spatial biomarkers as well-designed signatures capture pathway and cell type-related gene expression with higher fidelity. For a slide, correlations from all pairwise combinations of signatures (N = 15225) (i.e. the collection of all colocalization features) are referred to as the ‘colocalization map’ from here on (Fig. 1a).
A survival analysis was performed to ask whether slide-level patterns in colocalization maps harbored survival signals that could not be captured by the mere magnitude of signature expression. Three L1-regularized Cox proportional hazards regression models were fit to address this question, and the ability of the models to predict survival was computed with concordance indices (c-index) (Fig. 5a) (Methods). The first model only used flattened colocalization features (MOSBY predictions, N = 15225). The second Cox model only used signature expression levels (N = 175) with the goal of assessing the survival predictive power of gene expression ‘magnitude’. The third model was a joint model combining all signatures but only lasso-selected colocalization features from the first model in order to prevent colocalization features from dominating the model. Each model was run across 10 cross-validation folds to optimize the shrinkage parameter and also obtain mean and standard error estimates for the relevant c-index (Methods).
This process was implemented separately for all tested TCGA indications, and c-index mean and standard error estimates were plotted (Fig. 5a). We observed that the joint (i.e. third) model had a higher c-index than the signature-only model in most indications. This finding revealed that slide-level spatial patterns, discovered with an inference engine such as MOSBY, have survival predictive power that could not be captured by gene expression alone. As MOSBY colocalization features are biologically interpretable, this opens the door to discovering potentially ‘actionable’ colocalization or spatial exclusion patterns that predict clinical outcomes. Moreover, the c-indices achieved in the joint model were highly competitive with or higher than those reported in the literature from end-to-end survival-trained multimodal models utilizing genomic, transcriptomic, and image datasets44. Of note, comparing colocalization-only and signature-only models, we also found that the total survival predictive power of slide-level spatial patterns was not as high as that of signature levels in most TCGA indications. Ovarian and rectal cancer results were an exception to this general pattern (Fig. 5a), suggesting spatial biomarkers discovered in these indications may have the greatest potential to lead to novel insights.
Colocalization maps enable discovery of biologically interpretable spatial biomarkers
We next investigated consistent spatial predictors of risk that were supported across multiple indications and also showed evidence of tumor specificity. In each indication, survival effects and tumor specificity of colocalization features were explored with two tests: 1) A univariate Cox regression model for survival, and 2) a Mann-Whitney test to compare tumor-normal levels of colocalization value. A potential spatial biomarker of risk was defined as a colocalization feature that was significantly associated with poor overall survival (p < 0.05) and also had elevated levels of colocalization in the tumor (adj. p < 0.05). Given these criteria, four colocalization features had evidence in four different TCGA indications to be a spatial biomarker of risk (Fig. 5b). Of these four, the colocalization between an ER stress signature (XBP1s targets ER1722) and a neurotransmitter signature was associated with poor survival and malignant state in colon adenocarcinoma, lung adenocarcinoma, liver hepatocellular and ovarian cancers (Fig. 5c, 5d). In an independent non-squamous lung cancer study involving both immune checkpoint blockade (atezolizumab) and chemotherapy arms (IMpower110), this colocalization feature was also found to be associated with poor survival in the chemotherapy, but not immunotherapy arm, suggesting higher relevance as a resistance factor in chemotherapy (Fig. 5e, 5f). Of note, the ER17 and neurotransmitter signatures were not individually found to be associated with risk in any of the four mentioned indications (Supplementary Fig. 5a, 5b). Visual inspection of WSIs indicated that the expression of ER17 and neurotransmitter signatures primarily came from the microenvironment (as opposed to tumor region) in the case of high colocalization (Supplementary Fig. 5c). In low colocalization cases, the neurotransmitter signature expression primarily came from the tumor region whereas the ER17 signature expression was again predominantly in the microenvironment (Supplementary Fig. 5d).
Focusing on colocalization features involving immune system signatures, the T effector cell vs Cysteine colocalization was identified as the most consistent spatial biomarker of risk in TCGA. This colocalization feature was associated with poor survival and also showed significant tumor enrichment in breast, squamous lung, and ovarian cancers (Fig. 6a, 6b). T effector and cysteine signatures were not individually found to be associated with risk in any of these indications (Supplementary Fig. 5a, 6b). Visual inspection of WSIs indicated that the expression of T effector and cysteine signatures primarily came from the microenvironment (as opposed to tumor region) in the case of high colocalization (Supplementary Fig. 6c). As high colocalization is a risk factor, this expression pattern may be suggestive of a cysteine-associated immunosuppressive TME. In low colocalization cases, the T effector signature expression primarily came from the microenvironment whereas the cysteine signature expression was predominantly in the tumor region (Supplementary Fig. 6d).
The strongest survival effect for T effector vs cysteine colocalization was observed in breast cancer, where we investigated other immune cell types and found that immune vs cysteine colocalization was a general negative prognosis biomarker in this indication. Significant survival associations were observed for both lymphocytes/NK cells (Fig. 6c), and myeloid populations (Fig. 6d). The same immune vs cysteine colocalization features were also found to be negative prognostics in the Atezolizumab arm of Impower110 non-squamous cohort (Fig. 6e, 6f). Of note, most of these features were not prognostic in the chemotherapy arm (Supplementary Fig. 6e, 6f), however did not qualify as predictive biomarkers since the survival association differences between Atezolizumab and chemotherapy arms were not significant.
{kind=link}