2.1 Ischemia‒reperfusion, especially glucose deprivation-reperfusion, increases intracellular labile iron in renal tubular cells
As cells respond differently to IRI, we examined the effects of IR in multiple cell types including the human proximal tubular cell line Human Kidney-2 (HK2), the human renal proximal tubular epithelial cell line (RPTEC), and the cocker spaniel kidney epithelial cell line Madin Darby Canine Kidney (MDCK). These cell types were selected because kidney epithelial cells are among the main targets of IR that lead to loss of kidney function and play critical roles in the development of AKI, delayed graft function, and acute or chronic organ rejection in kidneys. To assess the effect of IR on cell labile iron, kidney epithelial cells were loaded with calcein-AM dye. The fluorescence started to quench after a 3-hour IR (not significantly); The fluorescence significantly decreased but still exhibited limited intensity after a 6-hour IR; and the quenching was almost complete after a 12-hour IR in these three different cells (Figure 1A). Calcein-AM fluorescence was quantified for individual cells after background subtraction and averaged. After 6 hours of IR, calcein-AM fluorescence decreased 72%, 78%, and 74% compared to that without IR. Meanwhile, after 12 hours of IR, fluorescence decreased 86%, 87%, and 84% in HK2, MDCK, and RPTEC cells, respectively (Figure 1B). To confirm that the cytosolic calcein quenching was attributed to an increase in labile iron, kidney epithelial cells were coincubated with 100 μM deferoxamine mesylate (DFOM) when performing 12-hour IR model. DFOM is a common iron chelator that chelates free Fe2+ but no other relevant cations that quench calcein-AM. DFOM strongly inhibited calcein quenching in cells treated with 12-hour IR, and the fluorescence level exhibited no sigfificant difference compared with that in control group cells (Figure 1A and 1B). Therefore, calcein quenching in kidney epithelial cells during IR signifies an increase in intracellular chelatable Fe2+.
IR injury can be divided into ischemia injury and reperfusion injury. Ischemic injury initiated by a lack of blood flow may cause hypoxia and nutrient deprivation. Reperfusion by blood flow causes secondary damage by inappropriate activation of ROS production and subsequent local inflammation. However, whether ischemia or reperfusion contributes to the increased labile iron during IR remains unknown. Therefore, we aimed to evaluate the impact of ischemia and reperfusion on LIP level by comparing the intracellular LIP levels of HK2 cells after ischemia alone or ischemia-reperfusion. Ischemic demonstrated significant increases in intracellular cell iron levels over normal cells, as evidenced by measurements of calcein-AM fluorescence. Reperfusion after ischemia led to further increase in LIP levels over ischemic only (Figure 1C and 1D). These results demonstrated that ischemia caused iron overload in kidney epithelial cells, and reperfusion after ischemia further exacerbated iron overload compared with ischemia alone.
The effects of kidney IR can be attributed to the effect of oxygen deprivation, serum deprivation and glucose deprivation. To investigate the influence of deprivation of these factors on iron overload, kidney epithelial cells were incubated under oxygen, glucose, or FBS (Fetal bovine serum) deprivation for 12 hours followed by reperfusion for 6 hours. During oxygen or FBS deprivation-reperfusion, virtually no quenching of calcein fluorescence was observed in the three types of kidney epithelial cells. In contrast, after GD-R, calcein-AM fluorescence was quenched in different cells (Figure 1E, 1F, S1). These results suggested that dysregulation of iron homeostasis after IR was linked to glucose deprivation rather than hypoxia or serum deprivation.
To investigate how glucose concentration alters the iron metabolism during IR, HK2 cells were cultured under different external glucose concentrations for 12 hours and reperfusion of glucose for 6 hours. Compared with the normal glucose concentration (25 mM), glucose concentration of 20, 15 or 10 mM in the medium did not affect the intracellular iron level significantly, while a significant decline in calcein fluorescence in HK2 cells was observed in glucose conditions of 5 mM or 0 mM (Figure 1G and 1H). These data showed that a low concentration of glucose and subsequent reperfusion also directly impacted the homeostasis of intracellular labile iron.
Energy substrates, particularly glucose, glutamine, and pyruvate, are used by cells to produce ATP. During ischemia, glucose metabolism is reduced and an exogenous supply of glutamine could fuel the Krebs cycle and contribute to the glutamate and succinate pools of cells. Under glucose-depleted culture conditions, a glutamine-dependent and glucose-independent tricarboxylic acid cycle may operate under aerobic or hypoxic conditions. In mitochondria-containing cells, pyruvate can enter the citric acid cycle within the mitochondrial matrix and undergo oxidative phosphorylation. In erythrocytes and oxygen-deprived tissue, pyruvate remains within the cytoplasm and is converted to lactate, a process known as anaerobic glycolysis. Therefore, glutamine and pyruvate can be used by cells to provide ATP. To confirm that the dysregulation of iron homeostasis induced by GD-R results from the depletion of energy, HK2 cells under GD-R were supplemented with glutamine or pyruvate. For comparison with the normal cultured HK2 cells, additional supplementation with pyruvate or glutamine did not alter the calcein fluorescence (Figure 1I). However, the quenched calcein fluorescence induced by GD-R was replenished by supplementation with glutamine or pyruvate (Figure 1I and 1J).
Our fluorescence microscopy data provides new findings for the changes in LIP levels in IR, namely that GD-R is the main factor inducing iron homeostasis dysregulation during the IR process, and this increase in LIP is related to energy deprivation. Energy metabolism substrates such as glutamine and pyruvate can restore energy supply and maintain LIP homeostasis.
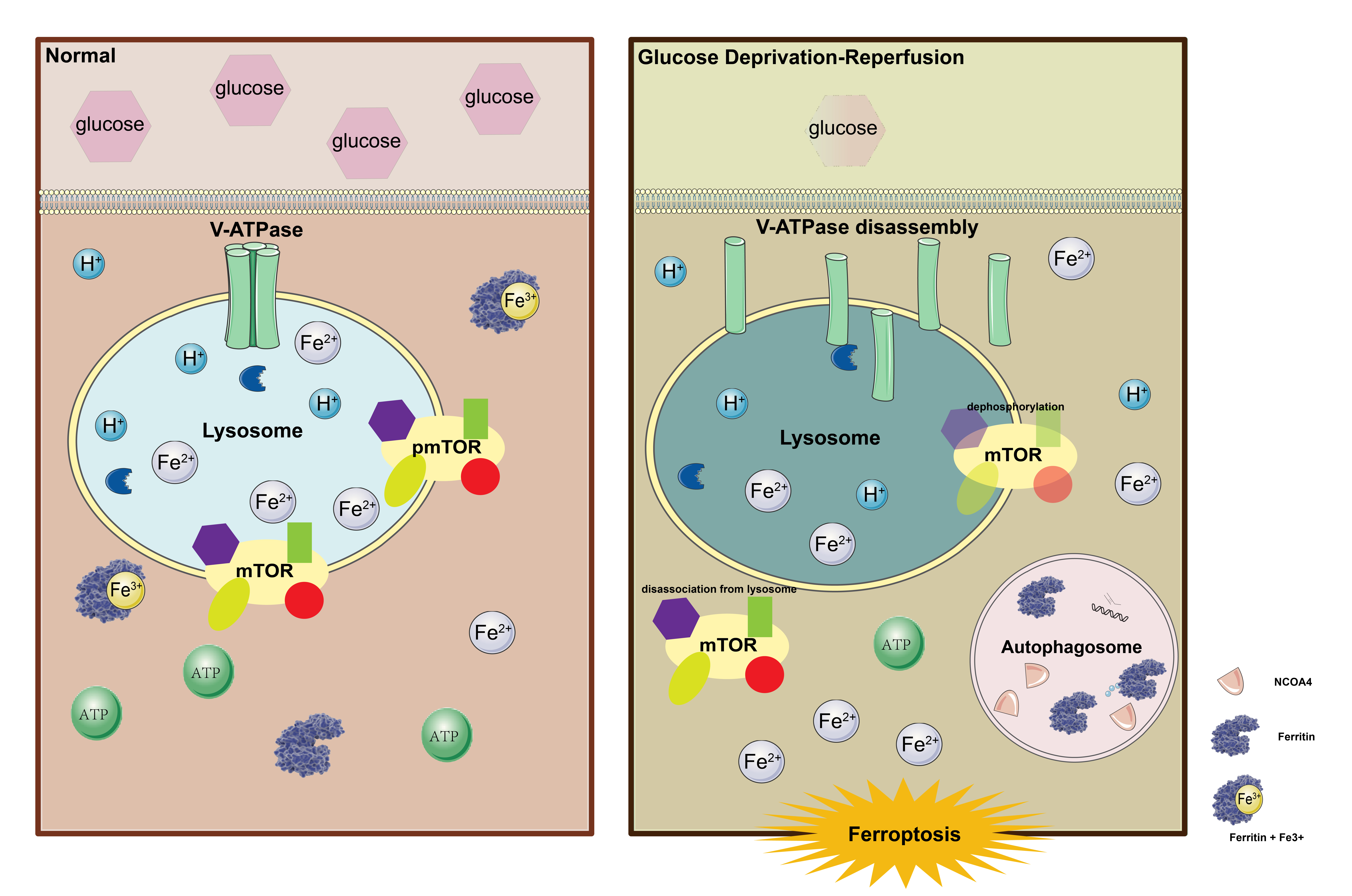
2.2 GD-R elevates lysosomal pH and inhibits the assembly and function of v-ATPase in renal tubular cells
Lysosomes play a key role in cellular and tissue health. Acidification contributes towards lysosomal function and the intracellular partitioning of metals, such as calcium and iron (22). To evaluation lysosomes, we first observed lysosomes in HK2 cells through transmission electron microscopy (TEM) to investigate the number of lysosomes in the cytoplasm of HK2 cells under the normal, IR, and GD-R conditions (Figure 2A). Compared with those in the normal group, HK2 cells in the IR group contained more lysosomes in the cytoplasm, and HK2 cells in the GD-R groups showed similar increases in the number of lysosomes. This result suggested that the degradation of lysosomes might be impaired during IR and GD-R, resulting in lysosome accumulation (23).
The alterations in lysosomal pH caused by IR were monitored by LysoSensorTM Yellow/Blue DND-160 staining in HK2 cells. Notably, only GD-R increased the lysosomal pH (Figure 2B and 2C), but neither oxygen nor FBS deprivation-reperfusion for 12 hours altered the lysosomal pH (Figure S2). The lysosomal pH increased with significantly enhanced blue fluorescence after exposure to GD-R for up to 6 and 12 hours; however, the change was not significantly after 3 hours of GD-R exposure (Figure S3). Additionally, the blue/yellow fluorescence ratio of the 12-hour group is higher than that of the 6-hour group, suggesting that GD-R induced the alkalization of lysosome in HK2 cells in a time-dependent manner. Meanwhile, the supplement of ATP inhibited the effect of GD-R on lysosomal pH, indicating that the increase of lysosomal pH may be due to the energy metabolism dysfunction induced by GD-R. These results suggested that GD-R plays a key role in lysosomal pH elevation in kidney epithelial cells.
V-ATPase is a membrane-bound, multi-subunit enzyme that pumps protons across membranes using the energy of ATP hydrolysis to maintain the lysosomal lumen at a pH of ~4.5 (24, 25). The function of V-ATPase is driven by ATP and regulated by glucose (26). Assembly of V-ATPase on the lysosomal surface is necessary for generating and maintaining the acidic environment within lysosomes, and the inactivation of V-ATPase is also one of the important reasons for the inhibition of lysosomal degradation (21, 27). The V1A subunit is located on the lysosomal membrane when V-ATPase was in assembly state and located in the cytoplasm when V-ATPase was disassembled. Therefore, to investigate the role of GD-R in the V-ATPase assembly in kidney epithelial cells, the amount of the V1A subunit of V-ATPase present in the membrane fraction and the whole cell was studied using western blotting. The V1A subunit levels in the cytoplasm were similar in HK2 cells under different treatment (Figure 2F and 2G). Notably, the V1A subunit levels in the membrane fraction of HK2 cells was significantly lower in the IR and GD-R groups than in the control group. The administration of ATP restored the membrane V1A subunit level after GD-R. In contrast, replenishment of 2-Deoxy-D-glucose (2-DG), which limits glucose uptake and metabolic pathways (abolish ATP generation), did not restore the membrane V1A level after GD-R (Figure 2D and 2E), suggesting that the inhibited assembly of v-ATPase induced by GD-R was based on impaired energy metabolism (28). Furthermore, to confirm this finding, a V-ATPase function assay was performed as described in the methods. Consistent with the western blot results, IR and GD-R inhibited V-ATPase function, which was restored by ATP, but not 2-DG (Figure 2H).
Next, to directly determine the relationship between LIP levels and V-ATPase function, we tested the labile iron in HK2 cells after GD-R, with or without carbamazepine (CBZ), a lysosomal acidification activator which can restore V-ATPase function (29). As expected, CBZ improved lysosomal acidification after GD-R, confirming the restoration of V-ATPase function after treatment of CBZ (Figure 2I and 2J). Consistent with the lysosomal pH levels, the increased labile iron under GD-R conditions was attenuated by CBZ, indicating that the GD-R induced labile iron increase is mediated by V-ATPase dysfunction.
Taken together, these results indicated that GD-R downregulates V-ATPase assembly and function in kidney epithelial cells through impaired energy metabolism, and the GD-R-induced V-ATPase dysfunction resulted in labile iron overload.
2.3 GD-R increases labile iron through V-ATPase-mTOR dysfunction and partially depends on TPC2
Researchers have established that V-ATPase is necessary for mTORC1 activation (30). However, whether the GD-R-induced V-ATPase dysfunction inhibits the mTORC1 pathway has rarely been reported. The phosphorylation of mTOR and S6 ribosomal protein is the hallmark of mTORC1 activation (31). Therefore, the phosphorylated and total contents of mTOR and S6 ribosomal protein in HK2 cells were analyzed to test the activation of mTORC1. We found that IR and GD-R did not affect the expression of total mTOR and S6, but they markedly decreased pmTOR/mTOR ratio and pS6/s6 ratio in HK2 cells (Figure 3A and 3B). Coculture with CBZ during GD-R significantly restored mTOR and S6 phosphorylation, indicating a significant relationship between mTOR activity and V-ATPase function. In addition, ATP also restored the proportion of pmTOR in HK2 cells after GD-R, while 2-dG did not, indicating the role of abnormal energy metabolism in mTOR dysfunction. Together, these results suggested that GD-R inhibited mTOR function in renal epithelial cells by inhibiting V-ATPase activity.
Meanwhile, the mTOR translocation to the lysosomal membrane is also an indicator of mTOR activation (32, 33), so we performed immunofluorescence assay of LAMP1 and mTOR to determine their localization. The IF images exhibited a slight but significant downregulation of mTOR accumulation in lysosomes after IR or GD-R (Figure S4). As expected, CBZ and ATP, but not 2-DG, restored the accumlation of mTOR in lysosomes under GD-R conditions, indicating that the dislocation of mTOR from lysosomes was attributed to the V-ATPase dysfunction and abnormal energy metabolism, verifying the immune blotting results.
Next, to further explore the underlying mechanism by which downregulated V-ATPase activity impacts the mTORC1 function, we investigated whether the GD-R-induced mTORC1 deactivation was associated with mTORC1 binding to V-ATPase, and would accompany GD-R-induced V-ATPase dysfunction. For this, we checked the presence of the mTOR in an IP against the V1 subunit V1A. The contents of V1A subunits and mTOR remained comparable in HK2 cells under GD-R conditions, but the mTOR content in the IP against V1A was decreased under GD-R condition, indicating the weakened binding between V1A and mTOR (Figure 3C). Furthermore, the restoration of V-ATPase function by CBZ increase the mTOR content in the IP during GD-R, indicating that the weakened binding was attributed to the inhibited V-ATPase function. Supplement of ATP also improved the binding, confirming the role of energy metabolism in GD-R-induced weakened binding between mTOR and V-ATPase.
Next, we investigated the role of mTOR in labile iron overload during GD-R. MHY1485, a potent mTOR activator, did not alter the cytoplasmic labile iron content under normal conditions while inhibiting labile iron overload after GD-R, while the rapamycin, a common mTORC1 inhibitor, simulated the effect of GD-R on labile iron. These results suggested that the GD-R-induced labile iron overload is dependent on the inhibition of mTOR (Figure 3D, 3E). In addition, treatment with MHY1485 during GD-R did not prevent the deacidification after GD-R in HK2 cells, indicating that the mTOR dysfunction might be a downstream target of V-ATPase under GD-R conditions (Figure 3F, 3G).
Endolysosomes contain a variety of cation channels, including two-pore segment channels (TPCs) (34). TPC2 is a ubiquitously expressed, lysosomally targeted ion channel that is inhibited after its associating with mechanistic target of mTOR (35). TPC2 activation is associated with increased cytosolic iron, indicating that these channels mediate lysosomal iron release (36). Therefore, we established TPC2 knockdown (KD) HK2 cells to determine whether the increased labile iron during the GD-R in renal epithelial cells is related to the function of TPC2 (Figure S5).
Compared with wt HK2 cells, tpc2 KD HK2 cells showed no differences in labile iron or lysosomal acidity under normal culture conditions. However, the cytoplasmic labile iron overload is significantly alleviated in tpc2 KD cells after GD-R or rapamycin treament (Figure 3D, 3E), indicating that mTOR-induced LIP overload is at least partially dependent on TPC2.
Taken together, our results indicated that GD-R inhibits mTORC1 activity by inhibiting the binding of mTOR to V-ATPase, and the mTORC1 dysfunction leads to LIP overload, which is partially attributed to the activation of TPC2.
2.4 GD-R-induced ferritinophagy plays a crucial role in iron metabolism disruption in kidney epithelial cells
Nuclear receptor coactivator 4 (NCOA4)-mediated ferritinophagy, which degrades ferritin, is a feedback regulatory mechanism for the available iron in cells (37). Ferritinophagy is among the autophagy subtypes and is an important mechanism by which cells can release iron from ferritin stores (38). Recent pathogenesis research on IRI indicates that IR induces autophagic degradation of ferritin through NCOA4, leading to labile iron overload (39). To further validate the relationship between ferritinophagy and GD-R, we analyzed the protein level of Microtubule-Associated Protein 1 Light Chain 3-B (LC-3B, a marker of autophagy) by fluorescence staining. The results showed an increased number of endogenous LC-3B in HK2 cells after 12 hours of GD-R, while treatment with ATP, 3-MA (an autophagy inhibitor) or MHY1485 inhibited the increase in LC-3B after GD-R (Figure 4A and 4B). Similar to LC3B level changes in HK2 cells, another mammalian autophagy protein Beclin1 was significantly upregulated after GD-R, while treatment with 3-MA or MHY1485 inhibited the GD-R-induced elevated Beclin1. Furthermore, we found that after GD-R treatment, the NCOA4 level was increased and ferritin was consequently decreased significantly in HK2 cells, which could also be prevented by ATP, 3-MA, and MHY1485 (Figure 4C and 4D). These data indicated that GD-R induced ferritinophagy in kidney epithelial cells, which results from energy depletion and the deactivation of mTOR.
To further confirm that ferritinophagy elevates labile iron levels and is regulated by NCOA4 during GD-R, ncoa4 KD HK2 cells were established (Figure S6). Compared with wild-type (WT) HK2 cells, ncoa4 KD did not change the ferritin level significantly under normal conditions. However, under GD-R treatment, a decrease in ferritin expression was observed in wt HK2 cells (Figure 23A, 23B), which was significantly inhibited in NCOA4 KD cells, indicating that the activation of NCOA4 mediated GD-R-induced ferritin degradation. (Figure 4E and 4F).
Since iron homeostasis can be regulated by NCOA4-mediated ferritinophagy, we aimed to test whether NCOA4 also mediated GD-R-induced intracellular labile iron overload. As expected, ncoa4 KD in HK2 cells did not alter the labile iron level under normal glucose supplement condition. However, compared with the high labile iron level in wt HK2 cells under GD-R conditions, the labile iron overload was significantly alleviated in ncoa4 KD HK2 cells (Figure 4G and 4H). Collectively, these results suggest that ferritinophagy plays a crucial role in GD-R-induced iron metabolism dysfunction in kidney epithelial cells.
2.5 GD-R-induced iron metabolism dysfunction results in ferroptosis in kidney epithelial cells.
Ferroptosis, a form of regulated cell death, is driven by iron-dependent lipid peroxidation and subsequent membrane destruction (40, 41). Ferroptosis is functionally relevant in the IRI of kidneys and other organs (42). The traditional belief was that a sudden increase in ROS production was induced by restored blood flow at early stage of reperfusion, and the burst of ROS is the main cause of IRI. Excessive ROS accumulation causes membrane lipid peroxidation, leading to ferroptosis (43). Nevertheless, limited studies were performed on iron metabolism dysfunction and its mechanism in IR injury, as well as the impact of iron metabolism on cell ferroptosis.
GD-R with different GD durations and the same reperfusion duration (6 hours) was performed to determine the association between the durations of GD-R and cell viability. GD-R with a GD duration longer than 6 hours inhibited cell viability in a time-dependent manner, while 3 hours of GD-R caused no significant inhibitory effects. Notably, coincubation with ATP or ferroptosis inhibitor ferrostatin-1 (Fer-1) effectively partially alleviated the loss of cell viability (Figure 5A). These results indicated that GD-R induced a decrease in cell viability in renal epithelial cells, which is associated with ferroptosis and energy depletion.
Ferroptosis is characterized by cellular oxidative stress, so we performed biochemical assays of intracellular redox indicators to further confirm the involvement of ferroptosis in GD-R-induced cell viability reduction. Compared with control group, GD-R resulted in excessive lipid peroxidation by-products malondialdehyde (MDA), and decreases in glutathione (GSH) and superoxide dismutase (SOD) (Figure 5B, 5C and 5D). Lipid peroxides, an important indicator of ferroptosis, were detected with Liperfluo probes. Our results showed that GD-R increased cellular lipid peroxides in a time-dependent manner (Figure 5E and 5F). Remarkably, these effects were significantly inhibited with Fer-1 or ATP (Figure 5B, 5C, 5D, 5E and 5F). Taken together, we confirmed that ferroptosis was implicated in GD-R-induced kidney epithelial injury, and energy depletion is the key mechanism that underlies ferroptosis.
Ferroptosis is iron-dependent cell death, and our previous results demonstrated that the mTOR signaling pathway, TPC2 and ferritinophagy were involved in the pathogenesis of GD-R-induced iron overload in kidney epithelial cells. Therefore, the iron chelator DFOM, mTOR activator MHY1485, tpc2 KD and ncoa4 KD HK2 cells were employed to explore the relationship among these mechanisms. Treatment with DFOM or MHY1485 significantly increased the viability of HK2 cells after GD-R compared with GD-R-induce HK2 cells. In addition, the viability of tpc2 KD or ncoa4 KD HK2 cells was significantly higher than that of WT HK2 cells after GD-R (Figure 5G). The treatment which promoted the iron homeostasis also improved the cell viability, indicating the role of iron metabolism in GD-R-induced kidney epithelial cell injury.
Correspondingly, MDA concentrations were significantly lower under DFOM or MHY1485 treatment, as well as in tpc2 KD or ncoa4 KD HK2 cells, than those in WT HK2 cells under GD-R conditions. In contrast, the levels of GSH and SOD in these groups were significantly higher (Figure 5H, 5I and 5J), indicating the ameliorative redox status. Furthermore, the Liperfluo fluorescence variation trends were similar to those of MDA among the different groups (Figure 5K and 5L), suggesting that the lipid peroxides were decreased with the improvement of labile iron overload.
Taken together, these results indicated that ferroptosis induced by GD-R in kidney epithelial cells was iron-dependent and could be alleviated by iron chelation, activation of mTOR or knockdown of TPC2 and NCOA4.
2.6 Ischemia-reperfusion induces iron overload and ferroptosis in mouse kidneys
In vitro results may not reflect in vivo conditions, so we further conducted in vivo tests to confirm our findings. To identify whether iron was overloaded and ferroptosis was activated in IR kidneys, we established uIRIx kidney model by ischemia for 15, 30, or 45 minutes followed by 48 hours of reperfusion with contralateral nephrectomy based on previous studies (Figure 6A) (44).
Consistent with the in vitro results, with the prolongation of ischemia time, the Fe(ii) contents in mouse kidneys increased significantly in the 30- or 45-minutes group (Figure 6B), as detected by colorimetric iron assay kits. These results indicated that iron overload occurred in uIRIx mouse kidneys.
Next, we measured renal function, morphology, oxidative stress, and lipid peroxidation in uIRIx mice with or without ferroptosis inhibitor Fer-1 to further investigate the association between renal IRI and ferroptosis. With prolongation of ischemia time and subsequent reperfusion, serum creatinine in mice were elevated severely and gradually. Moreover, treatment with Fer-1 significantly reduced the elevated blood creatinine in uIRIx mice, suggesting that ferroptosis might be involved in renal IRI (Figure 6C). Histological examination of renal tissues using hematoxylin and eosin (HE) and periodic Acid-Schiff (PAS) staining revealed that IR-induced tubular injury. 45-minute ischemia and 48-hour reperfusion resulted in obvious tubular damage, including tubular necrosis, tubular distension, and a loss of brush border membranes (Figure 6D and 6E). Additionally, Fer-1 treatment mitigated IR-induced tubular morphological injury, indicating that ferroptosis is involved in IR-induced tubular structural damage. Next, the levels of SOD, MDA, and GSH in uIRIx kidneys were evaluated through biochemical tests to determine renal oxidative stress reflecting ferroptosis. SOD and GSH levels in renal tissue were significantly decreased and MDA levels were significantly increased in uIRIx mouse kidneys compared with those in sham mice, and treatment with Fer-1 mitigated the changes in redox status (Figure 6F, 6G and 6H). Furthermore, immunohistochemical staining of 4-Hydroxynonenal (4-HNE), a key indicator for lipid peroxidation, was conducted to assess the lipid peroxides in uIRIx kidneys to confirm the IR-induced ferroptosis. Kidneys exhibited significantly more lipid peroxidation in 45-minute-IR group (Figure 6I and 6J), which was alleviated by Fer-1.
These results suggested that IR induces iron overload and ferroptosis in mouse kidneys. IR-induced ferroptosis is associated with kidney IRI, and inhibition of ferroptosis improves the function and morphology of mouse kidneys.
2.7 Iron chelators and mTOR activators alleviate IR-induced kidney injury by improving iron metabolism and inhibiting ferroptosis
To further identify the role of iron overload and mTOR signaling in ferroptosis and kidney injury induced by IR in vivo, mice after renal IR were subjected to DFOM or MHY1485 treatment. DFOM or MHY1485 administered by intraperitoneal injection significantly reduced the kidney iron content in uIRIx kidneys (Figure 7A), demonstrating that IR-induced iron overload in kidneys can be significantly inhibited by iron chelators or mTOR activators.
Next, the serum creatinine test showed that administration of DFOM or MHY1485 significantly improved the impaired uIRIx kidney function (Figure 7B), indicating that the reduction in iron contents mitigated the renal function damage. Consistently, the HE and PAS staining of mouse kidneys confirmed the alleviated tubular injury by DFOM and MHY1485 (Figure 7C). By further analyzing the biochemical characteristics of ferroptosis, we found that treatment with DFOM or MHY1485 significantly increased the SOD and GSH levels while inhibited the elevation of MDA in renal tissue after IRI (Figure 7D-7F). The 4-HNE staining also showed the alleviated lipid peroxidation induced by DFOM and MHY1485 (Figure 7G, 7H). Based on the serological, histological, biochemical, and IHC results, we revealed that ferroptosis and kidney injury induced by IR can be alleviated through iron chelators or mTOR activation.
To verify the biological relevance between iron overload and mTORC1 activation and explore its role in mouse renal ferroptosis after IR, the phosphorylation of mTOR and S6 in mouse kidneys was analyzed. Our results showed that the phosphorylated mTOR and S6 proteins were significantly decreased after IR, consistent with in vitro results. Additionally, the phosphorylation was restored in uIRIx kidneys with the treatment of MHY1485, while DFOM showed no significant effects on mTOR or S6 phosphorylation (Figure 7I, 7J).
To further elucidate the mechanism by which mTORC1 activation inhibits renal ferroptosis after IR, we analyzed tthe biomarkers of ferritinophagy. We found that renal IR significantly upregulated LC3B and Beclin1 protein expression in kidney tissue, suggesting the activated autophagy. Treatment with DFOM did not affect the level of LC3B or Beclin1 protein, but administration of MHY1485 significantly inhibited the elevation of LC3B and Beclin1 level (Figure 7K-7N), suggesting that activation of mTOR generates considerable effects on the prevention of autophagy in kidney epithelial cells. In addition, NCOA4 protein was increased and ferritin was decreased significantly in uIRIx kidneys, with or without administration of DFOM. As expected, increased NCOA4 and decreased ferritin were significantly inhibited by MHY1485 (Figure 7M, 7N). These changes in NCOA4 and ferritin expression were confirmed with IHC staining (Figure S7), indicating that IR induced ferritinophagy, which could be inhibited by mTOR activation. These results indicated that DFOM mitigated IR-induced ferroptosis by directly chelating iron in the cytoplasm, while MHY1485 improved iron metabolism by activating mTORC1 and inhibiting ferritinophagy.
Taken together, our in vivo results demonstrated that IR-induced ferroptosis in kidneys resulted from energy deprivation-mTOR inhibition-labile iron overload, and chelating iron and activating mTOR could alleviate iron overload and ferroptosis.
{kind=link}