General. Commercial reagents, standards, and solvents were purchased from Sigma Aldrich (Shanghai, China), Meryer Chemicals (Shanghai, China), Aladdin Reagents (Shanghai, China), J&K Chemicals (Beijing, China), and TCI Chemicals (Shanghai, China), and used without further purification.
Fermentation medium and conditions.A single colony of recombinant E. coli strain was cultivated overnight (10-12 h, 37 ℃) in LB medium (10 g·L-1 peptone, 5 g·L-1 yeast extract, and 10 g·L-1 NaCl; pH 7.0) with appropriate antibiotics (50 μg·mL-1 kanamycin) and used as the inoculum (1%). The culture was then transferred into 200 mL 2×YT medium (10 g·L-1 yeast extract, 16 g·L-1 peptone, 5 g·L-1 NaCl; pH 7.0) containing appropriate antibiotics in a 500 ml flask. When the OD600 of the culture broth reached 0.6-0.8, isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to a final concentration of 0.2 mM to induce gene expression. The cells were inducted at 16 °C for 16 h and collected by centrifugation (8000×g, 10 min).
Enzyme purification. The cell pellet was resuspended in 30 ml washing buffer (50 mM Na2HPO4, 150 mM NaCl, pH 8.0) containing a protease inhibitor and subsequently lysed by high pressure homogenizer. The lysed cells were incubated with DNase I (final concentration 0.1 mg·ml−1) for 30 min at 4 °C. After removing cell debris by centrifugation (10,000 r.p.m., 30 min, 4 °C). The cleared lysate was loaded on a Strep-Tactin column (Strep-Tactin Superflow high capacity), incubated for 60 min at 4 °C, and the protein was purified according to the manufacturer’s guidelines. Proteins were desalted using 10DG desalting columns (Bio-Rad) with PBS pH 10.0 and analysed by SDS–PAGE. When it is necessary to perform protein crystallization experiments, the subsequent experiments were performed on an ÄKTA pure system (GE Healthcare) with a HisTrap HP column (5 ml, GE Healthcare). Protein concentration of purified enzyme was measured by detecting absorbance at 280 nm using a NanoPhotometer N50 spectrophotometer and taking into account the calculated extinction coefficients with the ExPASy ProtParam Tool.3. All purification operations were conducted at 4℃ when necessary.
HPLC analysis. Analysis of the concentration of adduct 3 was carried out using Waters Alliance e2695 HPLC (Waters Co., USA) with UV detector at 269 nm. Analysis of the concentration of adduct 3a-3e was carried out using Waters Alliance e2695 HPLC (Waters Co., USA) with UV detector at 254 nm. Column: Agilent SB-C18 (150×4.6 mm, 5 μm), mobile phase: acetonitrile: H2O (0.7% HAc) =30:70, flow rate: 1 mL·min-1, temperature: 25 ℃. Peaks were assigned by comparison to chemically synthesized standard.
The separation and purification of adduct 3 was carried out using preparative high performance liquid chromatography Waters 2545 Binary Gradient Module (Waters Co., USA) with a Waters 2767 Sample Manager, and the UV detector at 269 nm. Column: CST Daiso C18 (250×20mm, 10 μm, 12nm), mobile phase: acetonitrile: H2O (0.7% HAc) =30:70, flow rate: 10 mL·min-1, temperature: 25 ℃.
The stereoselectivity of MBH adduct 3 were determined using Waters Alliance e2695 HPLC (Waters Co., USA) with UV detector at 269 nm. The stereoselectivity of MBH adduct 3a-3e were determined using Waters Alliance e2695 HPLC (Waters Co., USA) with UV detector at 254 nm. Column: DAICEL CHIRALPAK IC (250×4.6 mm,5 μm), flow rate: 1 mL·min-1, temperature: 25℃, mobile phase: n-hexane/isopropanol=90/10 (v/v). Peaks were assigned by comparison to the results of BH32.1423.
GC and GC-MS analysis. The concentrations of 3 and 4 were determined by using a GC (GC-2030, SHIMADZU, Japan) with a column (SH-5, 15 m×0.25 mm×0.1 μm; SHIMADZU). The GC analysis conditions were as follows: injector 310°C, and oven 45°C, hold 2 min, 10°C min-1 to 280°C, hold 10 min.
The identification of adducts (3, 3a-3e) were determined by using a GC-MS (Trace1310-TSQ8000, Thermo Scientific, USA) with a column (TG-5 30 m×0.25 mm×0.25 μm). The GC analysis conditions were as follows: injector 270°C, ion source temperature 300°C, and oven 80°C, 15°C min-1 to 280°C, hold 8 min.
Activity assay. The activity of MBHase was measured by HPLC. The assay mixture contained 300 µL PBS buffer (pH 10.0), including 1 mM 2, 5 mM 1 and 30 μM purified protein. Reactions were conducted in triplicate and incubated at 25°C and 220 rpm for 10 h and started by addition of the enzyme solution. The reaction was terminated with equal volume of 1 M HCl, centrifuged at 12000 rpm for 10 min, filtered by 0.22 μm filter membrane, and then determined by HPLC. One unit of specific activity is defined as the amount of adduct 3 (in nmol) that can be produced by one unit of enzyme in per minute under standard conditions.
Kinetic characterization. Initial velocity (V0) versus [4-nitrobenzaldehyde] kinetic data were measured using strep-tagged purified enzyme (30 µM), a fixed concentration of 1 (5 mM) and varying concentrations of 2 (0.5–35 mM). Reactions were performed in PBS pH 10.0 with 3% methanol and were incubated at 25 °C with shaking (220 r.p.m.) for 12 h. V0 versus [2-cyclohexen-1-one] kinetic data were measured using a fixed concentration of 2 (4 mM) and varying concentrations of 1 (0.5–10 mM) using the enzyme concentrations and buffer conditions described above. Samples were quenched with 1 vol. of 1 M HCl and analysed by HPLC. The Km and kcat values were calculated by nonlinear regression according to the Michaelis–Menten equation using GraphPad Prism software.
Mass spectrometry. Purified protein samples were buffer-exchanged into PBS (pH 10.0) using a 10 k MWCO Vivaspin unit (Sartorius) and diluted to a final concentration of 0.2 μM and then add 0.2% acetic acid. MS was performed using LTQ-Orbitrap Velos system, mass range: 500-1500, max inject time: 10 ms, resolution: 30000, sheath gas flow rate: 30, aux gas flow rate: 5, sweep gas flowrate: 1, capliary temp: 275℃, S-Lens RF Level: 69%, flow rate: 2 μL·min−1, record: 10 min. The resulting multiply charged spectrum was analyzed and deconvoluted using Unidec software.
Biotransformation procedures. For the reduction reaction catalyzing by GkOYE: Reactions were performed in 300 μL PBS buffer (pH 7.4) with 5 mM 1, 1 mM NADPH, 100 μM purified protein and 1% methanol as cosolvent. Reactions were incubated at 25°C and 220 rpm for 8 h. The solution was extracted with the same volume ethyl acetate (EtOAc). The resulting solution was dried by Na2SO4 and filtered through 0.22 μm membrane filters. Then the yield determined via GC.
For the MBH reaction catalyzing by GkOYE and mutants: For the product 3, 3a-3e, reactions were performed in 300 μL PBS buffer (pH 10.0) with 5 mM 1,1a, 1 mM 2, 2a-2e, 100 μM purified protein and 3% methanol as cosolvent. Reactions were incubated at 25°C and 220 rpm for 40 h. The reaction was terminated with equal volume of 1 M HCl, centrifuged at 12000 rpm for 10 min, filtered by 0.22 μm filter membrane, and then determined by HPLC. For the detection of stereoselectivity of MBH adducts. The solution was extracted with the same volume ethyl acetate (EtOAc). The resulting solution was dried by Na2SO4 and filtered through 0.22 μm membrane filters. Then the stereoselectivity was determined via HPLC with chiral column.
All the experiments were carries out at least in duplicate. The above products were further identified by nuclear magnetic resonance (NMR) analysis and shown in Figure Supplementary Figure 8-12. The representative HPLC chromatograms are shown in Supplementary Figure 13-24. The representative GC-MS chromatograms are shown in Supplementary Figure 25-29.
Preparation of racemic products standards. 2-(hydroxy(4-nitrophenyl)methyl)cyclopent-2-en-1-one (3) was separation and purification using preparative high performance liquid chromatography refer to the HPLC analysis for the specific process. The spectral data are consistent with literature values42. 1H NMR (600 MHz, Chloroform-d) δ 8.19 (d, J = 8.4 Hz, 2H), 7.57 (d, J = 8.4 Hz, 2H), 7.31 (s, 1H), 5.66 (s, 1H), 3.74 (s, 1H), 2.66 - 2.59 (m, 2H), 2.52 - 2.42 (m, 2H). 13C NMR (151 MHz, Chloroform-d) δ 209.45, 160.07, 148.68, 147.58, 146.85, 127.21, 123.83, 69.02, 35.26, 26.96. MS (EI): m/z calcd for C12H11NO4: 233.07; found: 233.1.
General procedure for the preparation of racemic product standards (3a-3e) was refer to the literature23. Arylaldehyde (10 mmol, 1.0 equiv.), cyclic enone (10 mmol, 1.0 equiv.) and imidazole (10 mmol, 1.0 equiv.) were stirred in 1 M NaHCO3 (40 ml) and THF (10 ml) for 48 h at room temperature. The reaction was acidified with 1 M HCl and extracted with ethyl acetate (3 × 50 ml). The organic layer was dried over MgSO4 filtered, and the solvent was removed in vacuo to give the crude product. The crude product was purified by silica gel chromatography (2:1 cyclohexane:ethyl acetate).
4-(hydroxy(5-oxocyclopent-1-en-1-yl)methyl)benzonitrile (3a):The spectral data are consistent with literature values43. 1H NMR (600 MHz, Chloroform-d) δ 7.63 (d, J = 8.4 Hz, 2H), 7.51 (d, J = 8.4 Hz, 2H), 7.30 (t, J = 2.7 Hz, 1H), 5.60 (s, 1H), 3.70 (s, 1H), 2.64 - 2.56 (m, 2H), 2.49 - 2.41 (m, 2H). 13C NMR (151 MHz, Chloroform-d) δ 209.38, 159.90, 146.93, 146.78, 132.41, 127.09, 118.80, 111.64, 69.17, 35.25, 26.91. MS (EI): m/z calcd for C13H11NO2: 213.08; found: 213.1.
2-((4-chlorophenyl)(hydroxy)methyl)cyclopent-2-en-1-one (3b):The spectral data are consistent with literature values44. 1H NMR (600 MHz, Chloroform-d) δ 7.31 (s, 4H), 7.26 (t, J = 2.7 Hz, 1H), 5.51 (s, 1H), 2.62 - 2.55 (m, 2H), 2.49 - 2.39 (m, 2H). 13C NMR (151 MHz, Chloroform-d) δ 209.66, 159.63, 147.54, 139.97, 133.68, 128.74, 127.86, 69.28, 35.33, 26.80. MS (EI): m/z calcd for C12H11ClO2: 222.04; found: 222.07.
2-((4-bromophenyl)(hydroxy)methyl)cyclopent-2-en-1-one (3c):1H NMR (600 MHz, Chloroform-d) δ 7.51 (d, J = 8.4 Hz, 2H), 7.31 (s, 1H), 7.30 (d, J = 8.4 Hz, 2H), 5.56 (s, 1H), 2.66 - 2.62 (m, 2H), 2.53 – 2.48 (m, 2H). 13C NMR (151 MHz, Chloroform-d) δ 209.62, 159.67, 147.50, 140.51, 131.67, 128.18, 121.80, 69.25, 35.32, 26.80. MS (EI): m/z calcd for C12H11BrO2: 267.12; found: 266.97.
2-(hydroxy(4-methoxyphenyl)methyl)cyclopent-2-en-1-one (3d): 1H NMR (600 MHz, Chloroform-d) δ 7.31 – 7.28 (m, 3H), 6.89 – 6.85 (m, 2H), 5.49 (s, 1H), 3.78 (s, 3H), 3.44 (d, J = 4.0 Hz, 1H), 2.61 – 2.54 (m, 2H), 2.48 - 2.41 (m, 2H). 13C NMR (151 MHz, Chloroform-d) δ 209.71, 159.33, 159.22, 148.07, 133.68, 127.78, 113.96, 69.62, 55.37, 35.38, 26.70. MS (EI): m/z calcd for C13H14O3: 218.09; found: 218.11.
2-(hydroxy(4-nitrophenyl)methyl)cyclohex-2-en-1-one (3e):The spectral data are consistent with literature values44. 1H NMR (600 MHz, Chloroform-d) δ 8.17 (d, J = 8.9 Hz, 2H), 7.53 (d, J = 8.9 Hz, 2H), 6.83 (td, J = 4.2, 1.1 Hz, 1H), 5.60 (s, 1H), 3.63 (s, 1H), 2.46 – 2.40 (m, 4H), 2.02 – 1.97 (m, 2H). 13C NMR (151 MHz, Chloroform-d) δ 200.17, 149.51, 148.27, 147.34, 140.36, 127.26, 123.63, 71.98, 38.53, 25.90, 22.49. MS (EI): m/z calcd for C13H13NO4: 247.08; found: 247.12.
Conservation analysis of the residues. Using The online Consurf Server (https://consurf.tau.ac.il/) and protein structure display software Pymol to analyze the residues conservation of GkOYE.
Measurement the volume of catalytic pocket. The volume of catalytic pockets of different mutants was measured using DoSiteScorer in the on-line software PROTEINS PLUS (https://proteins.plus/)45, 46
Crystallization, refinement and model building. All initial conditions of crystallization were screened using the sitting drop vapor diffusion method with Hampton Research Crystal Screen Kits. The crystal of the apo-GkOYE.8 was obtained by optimization after 2 days at 20°C in hanging-drop plates, under the conditions of mixing 0.8 μL protein solution (15 mg·mL-1) and an equal volume of reservoir solution (0.1 M glycine pH 9.0, 25% (w/v) polyethylene glycol 2000, 15% (w/v) glycerol). The crystal of the apo-GkOYE.8 is shown in Supplementary Figure 7. The crystals were cryoprotected by transient soaking in reservoir solutions with an additional 20–25% glycerol, and the crystals were flash cooled in liquid nitrogen at 100 K for data collection. X-ray diffraction data were collected on a Bruker D8 QUEST diffractometer (Karlsruhe, Germany), and the data sets were indexed, integrated and merged using XDS. The crystal structure of the apo-GkOYE.8 was solved by the single anomalous scattering method using a crystal that was diffracted to a 3.11 Å resolution. The structure of the apo-GkOYE.8 was solved using the molecular replacement method with a CCP4 automatic. The protein structure of 3gr7 was used as a searching model. Structural refinement was achieved using the Coot47 and Refmac548 programs. The data collection and refinement statistics of the apo-GkOYE.8 crystal structure are listed in Supplementary Table 2 and have been deposited in the PDB under accession code 8X0J. Structural figures were prepared using PyMOL v2.3.3 (Schrödinger, LLC, New York, USA).
The secondary structure of key mutants detected by circular dichroism. Preparation of protein: The purified target protein is tested by SDS-PAGE for protein purity. When the target protein content reaches 95% or above (the higher the protein purity, the more reliable the experimental results), the purified protein can be used for circular dichroism spectroscopy. Circular dichroic determination using JASCO-1700 circular dichroic spectrometer made in Japan, the spectra of 190-240 nm in the far ultraviolet were determined at room temperature. The concentration of the sample was 0.2 mg·mL-1 and the radius of the sample cup was 0.1 cm. The resolution is 0.5 nm, the bandwidth is 0.5 nm, the sensitivity is 50 mdeg, and the speed is 0.8 nm·min-1. 0.1*PBS buffer was used for the background solution. In the process of sample determination, 0.1*PBS buffer was first measured as the background, and then the mutant protein after buffer replacement. The resulting spectrogram data was analyzed by Dichroweb.
Library construction and screening. The primer sequences used to generate DNA libraries are shown in Supplementary Table 3. Site-directed saturation mutation was performed by PCR using mutagenic primers and plasmid pET28a-GkOYE.8 as template according to the manufacturer’s instructions of QuickChange (Stratagene). The DpnI-digested PCR product of 3 μl was used to transform 80 μl of E. coli BL21(DE3) chemically competent cells and colonies after transformation were incubated for DNA sequencing until all the designed mutants were obtained. Then refer to the Fermentation medium and conditions and Enzyme purification to obtained the pure protein of different mutants, and the conversion reaction was carried out with pure enzyme. HPLC was used to detect the product production.
Initial Structural Preparation for Computational Studies. The initial structure of GkOYE.8 were obtained by X-ray diffraction. The protonation states of the charged residues were determined at a constant pH of 10.0, based on pKa calculations via the H++ server (http://biophysics.cs.vt.edu/H++) and the consideration of the local hydrogen bonding network. In the GkOYE.7 models, residues His41, 44, 81, 95 and 167 were set as HIE, and residues His164, and 222 were set as HID. In the model, all Asp and Glu residues were deprotonated, while the Lys and Arg residues were protonated. The bond and angle force constants were determined using the Seminario method49, and point charge parameters for electrostatic potentials were determined using the ChgModB method. Each model was neutralized by the addition of Na+ ions and solvation in a truncated octahedral TIP3P water box with a buffer distance of 10 Å on each side. (R)-IntD was optimized at the B3LYP-D3/6-31 G(d,p) level by using Gaussian 16, the partial charge of these ligands was fitted with HF/6-31 G(d) calculations and the restrained electrostatic potential protocol50 implemented by the Antechamber module in the Amber 18 package. The force field parameters for these ligands were adapted from the standard general Amber force field 2.0 (gaff2)51 parameters, while the standard Amber19SB force field was applied to describe the protein.
Molecular Docking. To dock the (R)-IntD to the active sites of the GkOYE.8, 50000 uniformly distributed snapshots from the 100-ns MD simulation (with time intervals of 2 ps) were selected and divided into ten groups using a hierarchical agglomerative (bottom-up) approach. The optimized substrate (R)-IntD were docked to the active site of one representative group snapshot to mimic the ligand-protein complex. Molecular docking was performed with the Lamarckian genetic algorithm local search method using AutoDock Vina52. The docking approach was used for a rigid receptor conformation, while all rotatable torsion bonds of (R)-IntD were left free. A grid box was centered near the residues 26, 164, 167 and 169, and its size was set at 20 × 20 × 20 Å with a spacing of 0.375 Å. A total of 500 independent docking runs were performed with a maximum energy evaluation of 2.5×107. The 500 docked conformations obtained were clustered with an RMSD of 2.0 Å and ranked using an energy-based scoring function. The possible catalytically active binding modes were selected as initial configurations to perform MD simulations of GkOYE.8 in complex with (R)-IntD, according to the scoring function and reasonable conformation.
MD Simulations. All MD simulations were performed using the Amber 18 package software53. The MD pre-equilibrated GkOYE.8, and possible catalytically active binding modes of (R)-IntD was used as initial conformations for MD simulations of the protein-ligand complexes. Each system was brought to equilibrium with a series of minimizations interspersed by short MD simulations, during which restraints on the heavy atoms of the protein backbone were gradually released (with force constants of 10, 2, 0.1, and 0 kcal [mol Å-2]) and then slowly heated from 0 to 300 K for 50 ps. Finally, a standard unrestrained 100-ns MD simulation with periodic boundary conditions at 300 K and 1 atm was performed. The pressure was maintained at 1 atm and coupled with isotropic position scaling. The temperature was maintained at 300 K using the Berendsen thermostat. Long-range electrostatic interactions were treated using the particle mesh Ewald method, and a cutoff of 12 Å was applied to both particle mesh Ewald9 and van der Waals interactions54. A time step of 2 fs was used along with the SHAKE algorithm for hydrogen atoms, and a periodic boundary condition was used. For each system, total of three replicas of 100 ns each were carried out, accumulating a total of 300 ns of simulation time. The conformations visited by the enzyme along all this simulation time were clustered based on protein backbone RMSD, and the most populated cluster was selected as a representative structure of these enzymes. The CPPTRAJ module was used to calculate the stability (structure, energy, and temperature variations), convergence (RMSD of the structures), distance, and angle of each system in the AmberTools18 software53.
DFT calculations. DFT calculations were performed using the Gaussian 16 package. All DFT structures were constructed based on the catalytic mechanism55 and combined with the reaction conditions in this study. Geometry optimizations of the minima and transition states involved were performed at the B3LYP-D3 level of theory with the 6-31+G (d) basis set. Vibrational frequency calculations were performed at the same level to ensure that all stationary points were transition states (one imaginary frequency) or minima (no imaginary frequency) and to evaluate zero-point vibrational energies and thermal corrections at 298 K. Single-point energy calculations were performed at the B3LYP-D3 level using the 6-311+G(2d,p) basis set. Solvation by water was considered using the CPCM model56 for all of the above calculations. All Supporting computational data can be found in the Supporting Note2. Optimized DFT structures are illustrated with CYLView (https://www.cylview.org/).
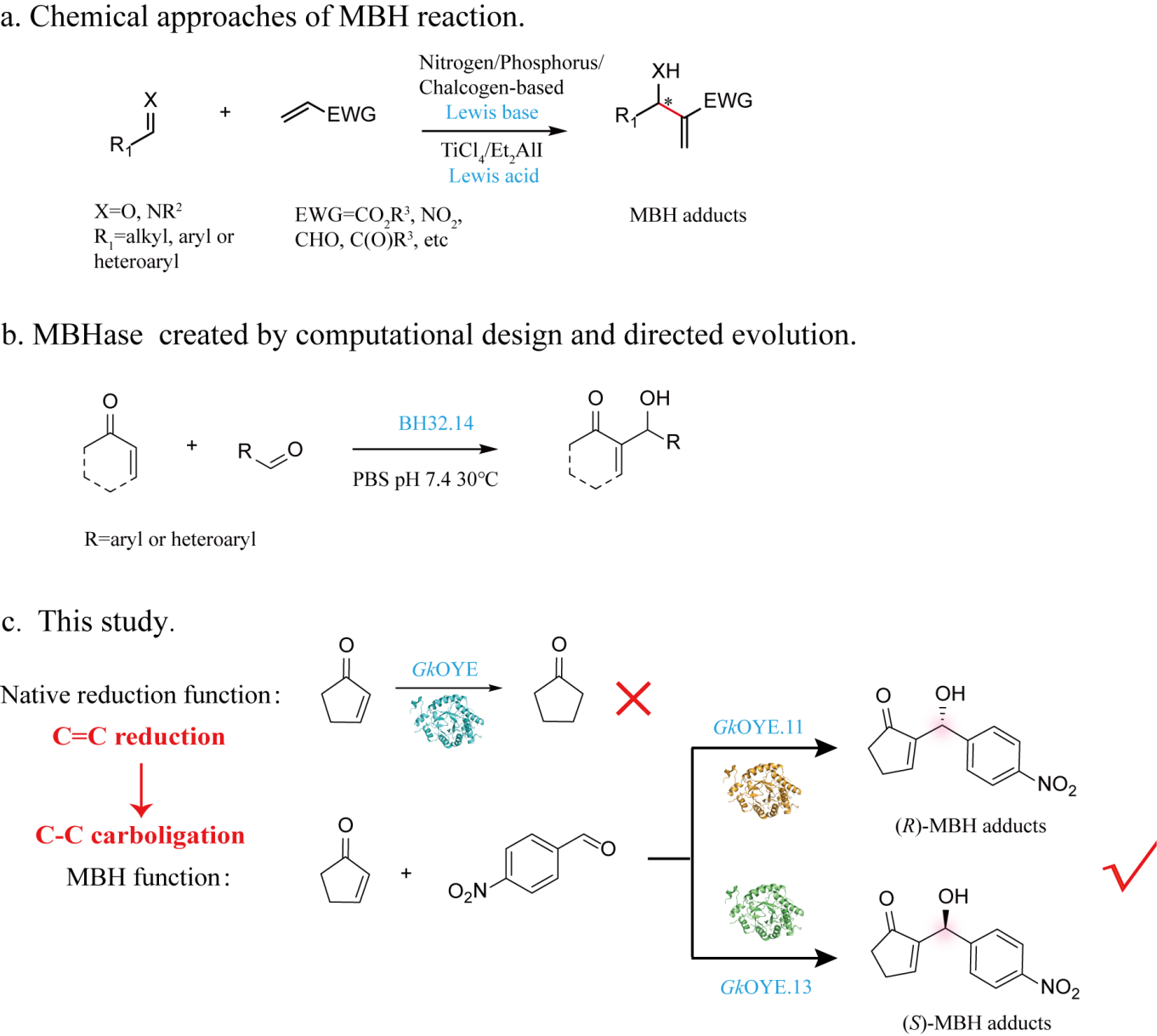{kind=link}