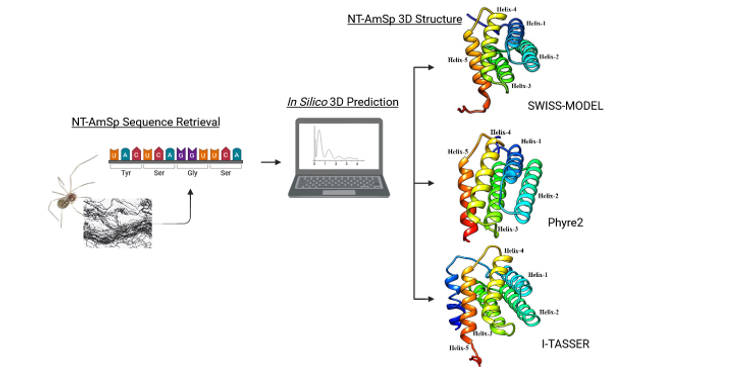
Many previous studies on the tertiary structure of spidroin N-terminal region have elucidated the insights of the domain properties and function [11, 37, 39]. Using the in silico approach, the tertiary structure of the monomeric N-terminal domain of the ampullate spidroin from C. lyoni prenymph was determined through computational tools including SWISS-MODEL, Phyre2, and I-TASSER, which are based on homology modeling, threading and ab initio methods. The structural prediction of NT-AmSp was performed on a monomeric basis rather than explicitly considering the homodimer state. Similar to other NT-domains of major ampullate spidroins, C. lyoni NT-AmSp was predicted to fold as an up-and-down (antiparallel) globular five-helix bundle (Fig. 2A, 2C, 2E) that conforms to the stereotypical spidroin tertiary structure of five helical domains [11, 13, 38, 40, 41]. As expected, all of the NT-AmSp predicted structures, threaded with high resolution, fit well with the solved MaSp1 from E. australis (4FBS), with RMSD scores ranging from 2.04 to 2.49. This result is consistent with the predicted structures of the end terminal domains of various spidroins, including the major ampullate, by homology-based prediction with RMSD scores between 0.38 and 2.5 [41]. The terminal domains, including the N-terminus, are evolutionarily conserved, suggesting a highly similar function across all spidroins [13]. Upon synthesis in the tail of the major ampullate silk gland, the spidroin is stored as a highly concentrated semi-crystalline liquid protein known as dope in the sac at physiological pH [42]. At neutral pH, the N-terminal domain adopts a monomeric state with five α-helical conformations, conferring solubility to the spidroin. A decrease in pH as the liquid protein travels down to the spinning duct causes the protonation of the carboxylate side chains, leading to the dimerization of the N-terminus with a pKa of around 6.5. This stabilizes the NT and pulls the spidroin into a tight network [43]. To date, the N-terminal of spidroin has been observed to remain highly soluble in its helical form under various conditions [42].
In water-soluble proteins, approximately 35% of the total amino acid residues adopt the α-helical conformation [44]. This characteristic was consistently observed in all predicted structures of NT-AmSp in the present study. SWISS-MODEL generated Model 1 of NT-AmSp, featuring five helical bundles that cover 61.9% of the total amino acids. The alignment with the homologous template, MaSp1A (7wio.1.A) from Triconephila clavipes [45], revealed the highest sequence similarity of 42.4% compared to four other models predicted by the server (Table 1). Phyre2 also predicted the top model, Model 1, with five α-helices covering 65.8% of the total residues. This prediction was based on alignment with MaSp1 (c3lr6A) from E. australis [11], sharing 44.0% similarity. Meanwhile, I-TASSER utilized PSI-BLAST to identify related sequences, with similarity percentages ranging from 29–44%. Subsequently, PSIPRED predicted secondary structures before threading the structures through the PDB structure library [23]. By integrating the ab initio method, Model 3 was generated, comprising a total of six α-helices covering 71.6% of the total residues. Notably, these included the same five helical bundles found in Model 1 by SWISS-MODEL and Phyre2. Surprisingly, the additional helical structure between Trp3 to Ala19, representing the putative signal peptide region, conformed to the typical structure of a signal peptide [46]. In many experimentally solved structures of MaSps using methods such as NMR and X-ray crystallography, the signal peptide region is often modelled as a random loop because the region is typically removed during protein translation in epithelial cells of the silk gland. It is worth noting that I-TASSER in this study stands out as a superior tool over homology and threading-based servers, providing extensive information on the NT-AmSp structure. Ab initio method is highly preferable when known template is unavailable [47].
Despite a high proportion of amino acid residues in the predicted α-helical conformations, not all of these helices are inherently hydrophilic, hydrophobic or amphipathic in nature. An α-helix represents the most common regular secondary structure in many water-soluble proteins [48]. Depending on the side chain chemical properties of amino acids, the α-helix can exhibit hydrophilicity or hydrophobicity with high number of polar residues or non-polar residues, respectively [49]. In some cases, it may display an amphipathic characteristic of being partially hydrophilic and hydrophobic [49]. However, to determine the nature of any of these helices in the predicted NT-AmSp, in-depth analysis is yet to be conducted. Nevertheless, the α-helices in the predicted NT-AmSp structures of all Model 1 and Model 3 were generated within similar amino acid regions, with only slight variations in the number of covered residues across the helix.
The predicted structure of the five α-helices of NT-AmSp are mainly hydrophobic. Helix-1 spans a range of 17 to 24 residues, with a high hydrophobic amino acid proportion of 66.7%, 58.3%, and 75.0% in Model 1 by SWISS-MODEL and Phyre2 and Model 3 by I-TASSER, respectively. Helix-2, on the other hand, was predicted in a 23-residual region with a high proportion of hydrophilic amino acids, constituting 65.2% in all models. Similar to Helix 1, Helix-3 possesses a considerable number of hydrophobic amino acids, ranging from 18 to 23 residues at 72.2%, 69.0%, and 71.4%, respectively. Interestingly, both Helix-4 and Helix-5 exhibit a mixture of hydrophobic and hydrophilic amino acids in relatively similar ratios, ranging from 47.0–55.0% and 45.0–52.9%, respectively. Both helical regions contain 17 to 23 amino acid residues in all models. Exclusively in Model 3, the α-helix core structure of the putative signal peptide region demonstrates an exceptionally long stretch of high hydrophobic amino acids, reaching up to 94.1%. It is a common feature found in all signal peptide motifs [50]. This result is corroborated by the previous hydropathy analysis by Kyte-Doolittle, showing that the N-terminal domain of AmSp is overall hydrophobic in nature, based on the calculated value of the grand average of hydropathicity (GRAVY) at 0.49 “(Mohtar), unpublished data)”. GRAVY measures the hydrophobicity of protein in which positive and negative values indicate hydrophobicity and hydrophilicity [51]. Similarly, the N-terminal domain of several spidroin types including tubuliform (TuSp) and pyriform (PySp) were also shown to display high amplitude of hydrophobicity [52, 53]. Nevertheless, the analysis was unable to determine the percentage of residues buried in the hydrophobic core or hydrophilic residues exposed on the surface of the NT-AmSp (Fig. 2B, 2D, 2F), which may contribute to understanding the orientation of the α-helices. The nature of the helices will remain vague unless determined experimentally.
From the present study, it can be inferred that the tertiary structure of the N-terminal ampullate spidroin from non-orbicularian species at an early developmental stage is highly conserved with that of orbicularian spiders in the adult stage. It is important to note that while these computational approaches provide valuable insights, the predictions are static and do not explicitly account for the protein being in a specific solution state, potentially limiting consideration of dynamic changes that could occur in a solution environment. In silico prediction tools with diversified algorithms, as used in this study, have proven beneficial as a preliminary window to provide insights into the three-dimensional structure of NT-AmSp from C. lyoni prenymph, which can be readily validated through advanced experimental methods such as Nuclear Magnetic Resonance (NMR) or X-ray Crystallography. With this information in hand, the predicted NT-AmSp structure can be employed to understand the functional diversity of the N-terminal domain of spidroin and utilized for the development of a soluble tag for in vitro or in vivo recombinant protein production [54].
{kind=link}