Catalyst characterisation
Ni/CeO2-NaNaph was prepared as described in our previous report with some modifications45,46. Briefly, a CeO2-supported Ni hydroxide precursor (Ni(OH)x/CeO2) prepared via deposition–precipitation was reduced by sodium naphthalenide (NaNaph) in tetrahydrofuran (THF) at room temperature (~24 °C) in an Ar atmosphere to afford Ni/CeO2-NaNaph (Ni: 1.38 wt%). The Ni K-edge X-ray absorption near-edge structure (XANES) spectrum of Ni/CeO2-NaNaph was between those of Ni foil and Ni(OH)2, which indicates that most Ni species in Ni/CeO2-NaNaph were zero-valent. According to linear combination fitting (LCF), ~76% of the Ni species were zero-valent (Fig. 2a; Supplementary Fig. 1a). This is mostly consistent with the X-ray photoelectron spectroscopy (XPS) results (Fig. 2b; Supplementary Fig. 2, Supplementary Table 1). The k3-weighted Fourier-transformed Ni K-edge-extended X-ray absorption fine-structure (EXAFS) spectrum of Ni/CeO2-NaNaph exhibited scattering originated from Ni−Ni bond of Ni metal in the first coordination sphere while very weak counterparts in the second coordination sphere were observed, suggesting the presence of highly dispersed Ni(0) species in Ni/CeO2-NaNaph (Fig. 2c; Supplementary Fig. 3, Supplementary Table 2). In fact, the diffuse reflectance infrared Fourier transform (DRIFT) spectrum of CO adsorbed on Ni/CeO2-NaNaph exhibited a peak around 2050 cm−1, which could be assigned to linear CO species adsorbed on zero-valent Ni species, but bridged species were not observed (Fig. 2d)47. Although direct observation of Ni species supported on CeO2 by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was difficult (Fig. 2e), STEM-energy-dispersive spectroscopy (EDS) mapping confirmed that highly dispersed Ni species were supported on CeO2 (Fig. 2f, 2g). The X-ray diffraction (XRD) patterns of Ni/CeO2-NaNaph were comparable with those of CeO2 and Ni(OH)x/CeO2, which supports that Ni(0) species were successfully immobilised on CeO2 with the CeO2 structures intact (Fig. 2h).
Catalyst effect
We initially investigated the effect of supported nonprecious-metal nanoparticle catalysts on acceptorless dehydrogenative aromatisation using 4-methylcyclohexanone (1a) as a model substrate under the reaction conditions indicated in Table 1. With Ni/CeO2-0.3NaNaph (prepared in the same manner as in our previous report45 by using 0.3 equivalent of NaNaph to the amount of NaNaph used to prepare Ni/CeO2-NaNaph), the desired dehydrogenated product p-cresol (2a) was obtained with a 50% yield along with 2-methylated phenol (2a′) with a 2% yield (Table 1, entry 1) (2a′ formation mechanism was discussed in the Supplementary Information). Interestingly, other supported 3d-transition-metal nanoparticle catalysts including Cu, Co, Mn, Fe and Zn did not catalyse dehydrogenative aromatisation (Table 1, entries 2–6). The Ni loading did not greatly affect dehydrogenative aromatisation (Supplementary Table 3). However, the yield of 2a was slightly improved by increasing the amount of reducing reagent to prepare Ni/CeO2-NaNaph (Table 1, entry 7). Next, we investigated the effect of the supports at 180°C. Only Ni/CeO2-NaNaph exhibited a high catalytic performance to afford 2a with a 74% yield (Table 1, entry 8). Ni catalysts on other supports such as hydroxyapatite (HAP), ZrO2, TiO2 and Al2O3 hardly catalysed the reaction (Table 1, entries 9–12). In addition, as-purchased CeO2, CeO2 reduced by NaNaph (CeO2-NaNaph) and Ni(OH)x/CeO2 did not catalyse the reaction (Table 1, entries 13–15). These results indicate that the reduced Ni was the actual active species. The solvent was also shown to have a substantial effect, and the yield of 2a was highest when N,N-dimethylacetamide (DMA) was used as the solvent (Supplementary Table 4). Dehydrogenative aromatisation of 3,5-dimethylcyclohexanone(1g) was immediately stopped by hot filtration of Ni/CeO2-NaNaph (Supplementary Fig. 4), and inductively coupled plasma atomic emission spectroscopy (ICP-AES) showed that almost no Ni species were in the filtrate (Ni: 0.21% of the Ni species used for the reaction), which indicates that the observed catalysis was heterogeneous.
To clarify the effect of the supports, Ni/CeO2-NaNaph was compared with Ni/HAP-NaNaph. The k3-weighted Ni K-edge EXAFS spectrum of Ni/CeO2-NaNaph exhibited very weak scattering originating from the Ni–Ni bond in the second coordination sphere whereas that of Ni/HAP-NaNaph exhibited relatively strong scattering (Supplementary Fig. 5, Supplementary Table 2). These results indicate the presence of ultrasmall Ni(0) nanospecies in the Ni/CeO2-NaNaph, which was likely due to the strong interaction between Ni(0) nanospecies and CeO245. Then, the effect of the basicity of the supports on dehydrogenative aromatisation was investigated. Dehydrogenative aromatisation of 1a was completely suppressed in the presence of benzoic acid (PhCOOH) (Table 1, entry 16). Meanwhile, dehydrogenative aromatisation of 1a using Ni/HAP-NaNaph was promoted by the addition of CeO2-NaNaph and Na2CO3, which indicates that the basicity of CeO2 promoted the deprotonative metalation step (Table 1, entries 17–19). Thus, the exceptional catalytic activity of Ni/CeO2-NaNaph for dehydrogenative aromatisation can be attributed to the high dispersion of Ni(0) species induced by strong interaction with CeO2 and the synergistic catalysis between Ni(0) species and the basic sites of CeO2.
Substrate scope
Next, we evaluated the substrate scope of Ni/CeO2-NaNaph for catalysing acceptorless dehydrogenative aromatisation of cyclohexanones (1) to phenols (2) (Fig. 3, 2a–2q from cyclohexanones; see Supplementary Table 5 for the detailed information on the by-products in substrate scope investigation). This catalytic system is applicable to para-, meta- and ortho-methyl-substituted phenol synthesis (1a–1c). Other alkyl-substituted cyclohexanones including 4-ethyl- (1d), 4-propyl- (1e), 3,4-dimethyl- (1f), 3,5-dimethyl- (1g) and 4-tert-butyl-(1h) ones were successfully converted into the corresponding phenols with good to moderate yields. This system could also be applied to the dehydrogenative aromatisation of aryl-substituted cyclohexanones (1i, 1j). In the case of cyclohexanones with various functional groups such as acetal, ethyl ester and acetamide, this catalytic system worked well at affording the desired dehydrogenated products with the functional groups intact (1k–1m). In addition, 1-tetralone (1n) and 1,4-cyclohexanedione (1o) were converted into corresponding phenols with good yields. Cyclohexenones such as 3-methyl-2-cyclohexen-1-one (1p) and carvone (1q) could be subjected to dehydrogenative aromatisation, but the C=C bond in the corresponding phenol obtained from carvone was hydrogenated.
Several studies have reported on the acceptorless dehydrogenation of secondary alcohols to afford the corresponding ketones in the presence of supported Ni nanoparticle catalysts48,49, which motivated us to investigate acceptorless dehydrogenative aromatisation of cyclohexanols (3) to phenols (2). Cyclohexanol is commercially obtained as ketone–alcohol (K–A) oil via hydrogenation of benzene followed by aerobic cyclohexane oxidation. Thus, if H2 formed by acceptorless dehydrogenative aromatisation of cyclohexanol is retrieved and used for benzene hydrogenation, then a formal process of aerobic benzene oxidation to phenol could be constructed21. As expected, 4-methylcyclohexanol (3a) was successfully converted into the corresponding phenol (2a) with a good yield in the presence of Ni/CeO2-NaNaph while a CeO2-supported Pd nanoparticle catalyst (Pd/CeO2-NaNaph) produced 2a with a much lower yield (Supplementary Table 6). This is because Pd nanoparticles hardly catalyse the dehydrogenation of cyclohexanols to cyclohexanones. The substrate scope of the catalytic system for dehydrogenative aromatisation starting from cyclohexanols (Fig. 3, 2a–2d, 2f–2h, 2n, 2o from cyclohexanols) includes various alkyl-substituted cyclohexanols (3a–3d, 3f–3h). In addition, 1,2,3,4-tetrahydro-1-naphthol (3n) and 1,4-cyclohexanediol (3o) were efficiently converted into phenols.
We found that Ni/CeO2-NaNaph could also catalyse the acceptorless dehydrogenative aromatisation of cyclohexylamines (4a–4d) to afford primary anilines (Fig. 3, 5a–5d), enamines (6a–6c) to afford aryl amines (Fig. 3, 7a–7d) and N-heterocycles (8a–8d) (Fig. 3, 9a–9d). With regard to dehydrogenative aromatisation for enamines, using Ni/CeO2-NaNaph to catalyse dehydrogenative aromatisation from 1-morpholino-cyclohexene (6a) resulted in the gradual formation of a dehydrogenated product (7a) via sequential dehydrogenation (Supplementary Fig. 6a) to achieve a high yield of 7a (Fig. 3), whereas using Pd/CeO2-NaNaph as the catalyst produced 7a and the hydrogenated product (7a′) at a ratio of 1:2 immediately after the reaction was started due to the disproportionation of 6a on Pd nanoparticles50 (Supplementary Fig. 6b), which resulted in a moderate yield of 7a even after 24 h (Supplementary Fig. 7). Then, Ni/CeO2-NaNaph-catalysed a,b-dehydrogenation of N-alkyl piperidones (10a–10c) was conducted (Fig. 3, 11a–11c), which surprisingly selectively converted 1-methyl-4-piperidone (10a) into 1-methyl-4(1H)-pyridinone (11a) with an excellent yield via double dehydrogenation of 10a. This reaction has not previously been reported. The reactions from 1-methyl-4-piperidone with a methyl group at the a-position of the carbonyl group (10b) and 1-ethyl-4-piperidone (10c) were successful, and they afforded corresponding double-dehydrogenated products with good yields.
Mechanistic studies
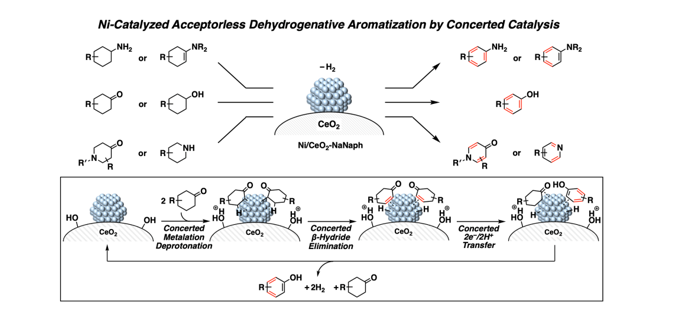
To confirm that the dehydrogenative aromatisation proceeded without any hydrogen acceptors, gas chromatography (GC) was used to analyse the gas phase after dehydrogenative aromatisation of 1g (Supplementary Fig. 8). The results showed approximately two equivalents of H2 to the dehydrogenated product (2g), which indicates that the dehydrogenative aromatisation was the acceptorless type. In addition, when 3-methyl-2-cyclohexen-1-one (1p) was the substrate, m-cresol (2b) and 3-methylcyclohexanone (1b) were produced with almost the same yields 10 min after the reaction was started (Supplementary Fig. 9). Together with the above results on support effects and our previous reports on acceptorless dehydrogenative aromatisation by supported Pd nanoparticle catalysts21–27,50,51, we propose the following mechanism for the formation of phenols from cyclohexanones (Supplementary Fig. 10): (1) cyclohexanones adsorb onto the surface of Ni nanoparticles; (2) cyclohexanones undergo concerted metalation deprotonation to Ni species with the assistance of Brønsted base sites in CeO2; (3) b-hydride elimination takes place to afford cyclohexenones; (4) cyclohexenones undergo disproportionation to afford phenols and cyclohexanones on the surface of the Ni nanoparticles and (5) H2 evolution occurs to achieve the catalytic cycle.
A series of kinetic analyses were conducted to determine the turnover-limiting step. We confirmed that the initial rate for the dehydrogenative aromatisation of cyclohexanone (1r) was saturated to the initial substrate concentrations under normal conditions in the present catalytic system (Supplementary Fig. 11), which indicates that the adsorption of 1r to Ni species was not the turnover-limiting step. The 2b production rate using 1p as the starting material was much higher than that using 1b (Supplementary Fig. 12), which indicates that the disproportionation of cyclohexenones was not the turnover-limiting step. The kinetic isotope effect (KIE) was not observed when 1r and cyclohexanone-a-d4 (1r-a-d4, kH/kD4 = 1.1) were used, which indicates that deprotonative metalation and H2 evolution were not the turnover-limiting steps (Fig. 4a). In contrast, KIE was observed when 1r-a-d4 and cyclohexanone-d10 (1r-d10, kD4/kD10 = 2.3) were used (Fig. 4a). These results indicate that the turnover-limiting step of Ni/CeO2-NaNaph-catalysed dehydrogenative aromatisation is b-hydride elimination. On the other hand, in the presence of Pd/CeO2-NaNaph, KIE was observed with 1r and 1r-a-d4 (kH/kD4 = 2.6) but not with 1r-a-d4 and 1r-d10 (kD4/kD10 = 1.3) (Fig. 4b), which indicates that the turnover-limiting step of Pd/CeO2-NaNaph-catalysed dehydrogenative aromatisation is the deprotonative metalation of cyclohexanones.
We conducted various control experiments to elucidate why our catalytic system enabled unprecedented Ni-catalysed acceptorless dehydrogenative aromatisation. First, we compared Ni/CeO2-NaNaph with other Ni complex catalysts for the dehydrogenative aromatisation of 1a (Supplementary Table 7). While 2a was not produced in the presence of Ni(II) complexes (Supplementary Table 7, entries 1–4), some 2a was produced when Ni(cod)2 and IPr (1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene) (Ni-IPr complex) were used (Table 1, entry 20). To clarify which steps were difficult for the Ni complexes, several experiments using deuterium-labeled cyclohexanones were conducted. The H/D exchange of 1r-a-d4 efficiently proceeded 12 h after dehydrogenative aromatisation was started in the presence of the Ni-IPr complex (Fig. 4c), whereas the use of either Ni(cod)2 or IPr caused the H/D exchange of 1r-a-d4 to hardly proceed under the same reaction conditions (Supplementary Fig. 13). Thus, a-C–H activation of 1r could occur with the Ni-IPr complex. However, the GC-MS and 2H NMR spectra of 1r-b,g-d6 (deuteration rate of a-position = 4%) after stirring with the Ni-IPr complex at 160°C for 12 h were comparable to those before stirring, which indicates that a decrease in D from 1r-b,g-d6 or H/D scrambling between the a- and b-positions via b-hydride elimination followed by insertion did not occur (Fig. 4d; Supplementary Figs. 14–16). These results indicate that b-hydride elimination was difficult to induce in the case of Ni complexes. Based on these results and the different turnover-limiting steps of Ni nanoparticles and Pd nanoparticles, we concluded that Ni species generally have difficulty with b-hydride elimination because of their smaller atomic radius compared with Pd species38–41, but the concerted catalysis by multiple active sites (Ni–Ni metal ensembles) of Ni nanoparticles could help attenuate the steric restriction and facilitate b-hydride elimination, which explains how our catalytic system achieved the unprecedented Ni-catalysed dehydrogenative aromatisation (Supplementary Fig. 17).
As noted earlier, when 1p was used as the starting material, fast disproportionation occurred to afford 2b and 1b in the presence of Ni/CeO2-NaNaph. However, the Ni-IPr complex hardly catalysed the reaction (Fig. 4e). Considering the different mechanisms of Pd mononuclear active sites and Pd nanoparticles for dehydrogenative aromatisation to phenols51,52, such a fast disproportionation of cyclohexenones to phenols and cyclohexanones appears specific to metal nanoparticle catalysts. This may be because the disproportionation proceeds via a concerted two-electron/two-proton transfer from one cyclohexenone to another cyclohexenone after the two cyclohexenone molecules adsorb together on metal nanoparticles with the metal ensembles (Supplementary Fig. 18). Furthermore, acceptorless dehydrogenation of 4,4-dimethylcyclohexanone (1s), which affords nonaromatic dehydrogenated products even after double dehydrogenation, did not proceed with Ni/CeO2-NaNaph, but small amounts of the corresponding dehydrogenated products 4,4-dimethylcyclohexa-2,5-dien-1-one (2s) and 4,4-dimethylcyclohex-2-en-1-one (2s′′) formed in the presence of Pd/CeO2-NaNaph (Fig. 4f). Based on the facileness of migratory insertion of Ni–H species to olefins compared with that of Pd–H species42–44, these results indicate that the re-insertion of Ni–H species produced by b-hydride elimination of 1s to 2s′′ occurs faster than H2 evolution, which explains the lack of dehydrogenated products from 1s with Ni/CeO2-NaNaph. Therefore, the fast disproportionation of cyclohexenones via the concerted two-electron/two-proton transfer on Ni nanoparticles to produce phenols, which are difficult to insert into Ni–H species, is another key factor for how we achieved Ni-catalysed acceptorless dehydrogenative aromatisation.
{kind=link}