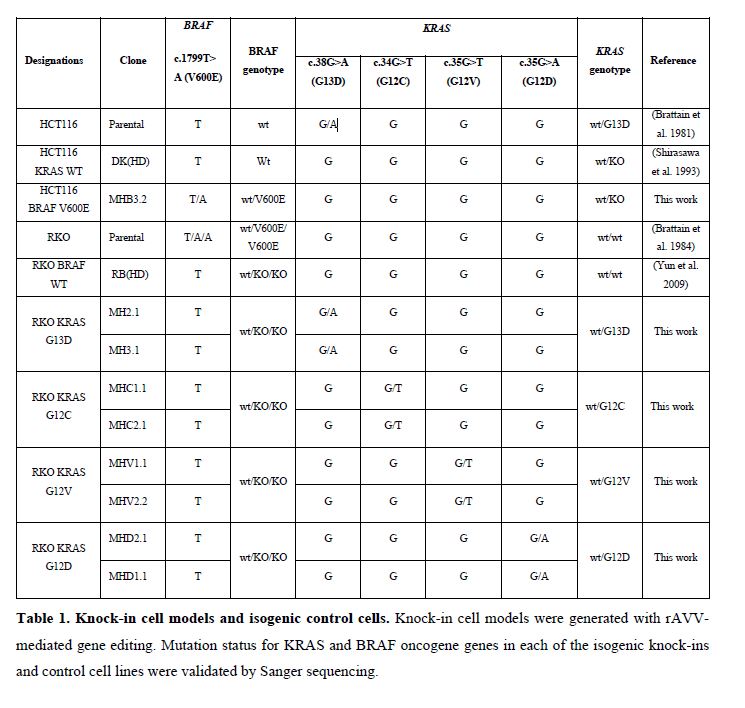
Generation of isogenic cell models of KRAS and BRAF mutations.
To better understand whether KRAS and BRAF mutations prevalent in CRC engender the same phenotype, we created a set of isogenic cell lines by introducing different mutant KRAS alleles in a genetic background where the mutant BRAF V600E allele had been removed by knock-out. Parental RKO cells [24] have two BRAF V600E mutant alleles and one wild-type allele, where the two BRAF V600E mutant alleles have been removed in RKO BRAF WT cells [5] whereas parental HCT116 cells [25] have a single KRAS G13D allele removed in HCT116 KRAS WT cells [26]. We used genome editing by recombinant adeno-associated virus (rAAV) technology [27] to knock in mutant KRAS and BRAF alleles in RKO BRAF WT and HCT116 KRAS WT cells, respectively. Gene targeting constructs were generated by amplifying homology arms from the respective targeted CRC cells followed by introduction the mutations by overlapping PCR [28] (Supplementary Fig. 1A). The KRAS mutant constructs were then used to target RKO BRAF WT cells, resulting in three independent edited clones of each KRAS genotype (Supplementary Fig. 1B). Presence of the desired KRAS mutation in targeted cells was demonstrated by sequencing the targeted KRAS exon and the expression of wild type and mutant alleles of KRAS and BRAF was confirmed by Sanger sequencing of the RT-PCR products (Supplementary Fig. 2A-B; Supplementary table 1). Finally, expression of the desired wild-type and mutant transcripts of KRAS and BRAF genes was confirmed by transcriptome sequencing (Supplementary Fig. 3A-G). Thus, a set of cell models where BRAF V600E and the KRAS codon 12 and 13 mutations can be studied in the same genetic background was generated and validated.
Transcriptomic, proteomic and metabolomic analyses of KRAS and BRAF mutations.
To find and understand common and distinct phenotypes of different Ras pathway mutations in CRC, we characterized the transcriptomes, proteomes and metabolomes of RKO and HCT116 cells with BRAF V600E mutation, KRAS mutations, or no Ras pathway mutation. At the transcriptome level, principal component analysis (PCA) of differentially expressed transcripts in the RNA sequencing data showed clear separation by genetic background between HCT116 and RKO cell lines and the derived cell clones. Surprisingly, no clear separation by Ras pathway mutation was observed (Fig. 1A). Similarly, PCA analysis of ~ 4,500 detected proteins separated the cell clones by genetic background but not by KRAS/BRAF mutation status (Fig. 1B). However, the metabolomics PCA analysis separated RKO as well as HCT116 cell clones by their Ras pathway genotype (Fig. 1C). The KRAS mutant cell lines clustered by mutation, separating mainly along the first principal component, which supports that the introduced mutations are responsible for more variation than any other variable. Taken together, under normal cell culture conditions the overall impact of the Ras pathway mutations at the transcriptome and proteome levels appeared limited whereas stronger effects were observed at the metabolome level.
Differential expression of genes and proteins in KRAS/BRAF mutant cells.
We next sought to determine whether the different KRAS and BRAF mutations alter the expression of specific genes or proteins. Because of the availability of multiple independent clones per genotype, we focused these analyses on the RKO genetic background. To find differentially expressed genes (DEGs) and proteins (DEPs) regulated by any or all of the different Ras pathway mutations, we compared their gene and protein expression data to the corresponding RKO isogenic controls (Supplementary Fig. 4A-E and 6A-D; Supplementary table 2A-J and 4A-I). We first identified 181 and 766 DEGs between all KRAS mutant clones and WT or BRAF V600E cells, respectively (Fig. 2A-B). For each KRAS mutation studied, the ratio of DEGs between comparisons to BRAF V600E or wildtype was in the range 1.90–2.49 (Supplementary table 2). The notion that the transcriptional response differs more between KRAS and BRAF mutant cells than between KRAS mutants and cells lacking Ras pathway mutation was supported by analyses of HCT116 cells, where the ratio was 1.56 (Supplementary Fig. 5). We hypothesized that the 22–34% of DEGs found both in comparisons with BRAF mutant and wild-type cells and having the same direction of expression change could be specific to a particular KRAS mutation (Supplementary Fig. 5A-D and F). We identified 35, 70, 26 and 39 such KRAS G12C/D/V/G13D mutation specific DEGs, respectively (Supplementary Fig. 5A-D and F; Supplementary Fig. 5E; Supplementary table 3). We next sought to determine whether these DEGs were also controlled by KRAS or BRAF mutations in clinical samples. Several DEGs were differentially expressed with the same direction of gene regulation in a KRAS G12 specific manner also in TCGA COAD data, including BCHE, BEST3, EXO1 [29], FCER2, FGF19, GPM6A, HOTAIR [29], KCNIP3, NTSR1, PRKAA2, SMC4, TMEM71, and TUBAL3 (Supplementary table 4A-J) where EXO1 and HOTAIR have previously been linked to Ras signaling. The expression of EXO1 and SMC4 is regulated by the DREAM complex [30], and they interact during DNA replication in yeast. Collectively, common and KRAS mutation specific DEGs with known as well as previously unknown links to the Ras pathway were identified.
Next, we proceeded to identify DEPs regulated by Ras pathway mutations. The ratio of DEPs identified in comparisons of KRAS mutants to BRAF V600E or wild-type was in the range 7.4–24 (Supplementary table 5). As compared to RKO cells with no Ras pathway mutation, known Ras regulated or interacting proteins were identified as DEPs: LGALS1 [31] was a DEP in KRAS G12D and G13D mutants, whereas IFI16 [32], S100A10 [33], CD44 [34], GLRX [35] and AHNAK2 [36] were DEPs in one of the KRAS mutants (Supplementary table 5B, D, F and H). From the proteins highlighted in [7], AKAP12 was a DEG in comparisons of KRAS mutant to BRAF V600E as well as wild-type cells but not a DEP. Interestingly, 6 DEPs were common to all four KRAS mutations when comparing to BRAF V600E, of which 3 were upregulated (LCP1, S100A10 and S100A2 [37]) and 3 downregulated (CRABP2 [38], FLNA [36] and NXN) more than 10-fold in KRAS mutant clones (Fig. 2C) (Supplementary Fig. 6A-D; Supplementary table 5A, C, E and G). When identifying KRAS G12C/D/V/13D mutation specific DEPs versus BRAF V600E, we identified 2 (OCRL and VAMP8), 3 (OCIAD2, H1-0 and S100A13), 7 (ANXA2 [39], GNG12, METTL7B, PROCR, CGB1, CD44 and CA9) and 7 (PHGDH [40], AHNAK2 [36], ASMTL, CPT1A, FASTKD5, HMGA1 [41] and FTH1), respectively. Notably, CD44, PROCR and HMGA1 have previously been found upregulated by KRAS G12V [42]. To assess the link between transcription and translation, we identified 11, 14, 17 and 21 DEGs, respectively, as regulated at both levels by KRAS G12C/D/V/G13D mutation as compared to BRAF V600E (p ≤ 3.58E-18, hypergeometric distribution; Fig. 3A-D). Of these, CRABP2, FLNA, LCP1, NXN, S100A2, and S100A10 were regulated at both levels in KRAS mutant cells. While the majority (61–78%) of DEPs in all four mutants were also DEGs, only 0.2–0.5% of the DEGs were also DEPs; the vast majority of transcriptional regulation was not reflected in altered protein expression while more than half of DEPs were regulated through altered gene expression. Thus, 6/6 of DEPs identified in comparisons of mutant KRAS clones to isogenic cells with no Ras pathway mutation have previously known roles in Ras signaling, whereas 6 common and 19 KRAS mutation specific DEPs of which 8 were previously known were identified in comparison to BRAF V600E isogenic cells.
Pathway analysis of transcriptome and proteome datasets.
Next, we sought to investigate common and unique pathways controlled by different KRAS mutations and whether different pathways are involved in KRAS and BRAF signaling through Ingenuity Pathway Analysis (IPA) of transcriptomes (Supplementary table 6A-D) and proteomes (Supplementary table 7A-B). In comparison to cells lacking Ras pathway mutation, BRCA1-DNA damage signaling, mismatch repair, and cell-cycle checkpoint signaling was enriched in KRAS G12D mutant cells (Supplementary table 6B) and actin-based motility, RhoGTPase signaling, and axonal guidance signaling in KRAS G13D (Supplementary table 6D) (Fig. 4A). Compared to BRAF V600E cells, the Integrin-Linked Kinase (ILK) pathway was significantly altered in KRAS G13D cells at the transcriptome and protein levels, as well as in KRAS G12D at the proteome level (Fig. 4B and C; Supplementary table 6D and 7B). The Molecule Activation Prediction (MAP) tool predicted ILK signal activation based on the protein expression changes in KRAS G12D and G13D, which is in turn predicted to activate downstream genes related to cell proliferation, adhesion, motility, cancer progression EMT, cancer stem cell markers and chemoresistance [43, 44] (Supplementary Fig. 7A-C). Additionally, a canonical pathway analysis of DEPs versus BRAF V600E showed enrichment of the Wnt/beta-catenin signaling pathway in KRAS G13D clones and of the serine biosynthetic pathway in G12D clones (Supplementary table 7A-B). Taken together, known (MMR, ILK, cell cycle checkpoint, actin-based motility, RhoGTPase signaling, axonal guidance, serine and glycine biosynthetic pathways) as well as novel (BRCA1-DNA) pathways were significantly regulated as a consequence of Ras mutation and the ILK pathway emerged as regulated by KRAS but not BRAF mutation.
Differential expression of immune related genes by Ras pathway mutations.
Co-expression of immune related genes and pathways has been used for immunological stratification of CRC [45, 46]. Several different immune regulatory pathways such as IFNα, IFNγ, host-defense and antigen presentation are suppressed in KRAS mutated CRC [47]. This prompted us to investigate the expression status of genes of three pathways in the different isogenic cell models: (i) the IFNα pathway (105 genes), (ii) the IFNγ pathway (149 genes), and (iii) KRAS mutually exclusive genes (56 genes) as defined in [47]. Several genes from these pathways were differentially expressed here in all the KRAS mutants (Supplementary Fig. 8A-C; Supplementary table 8A-C). These included BATF2, CCRL2, IFI27, IFI44L, SAMD9L, and TRIM21 from the IFNα pathway, and IRF9, MX2, and HLA-B from the IFNγ pathway which were DEGs in all the KRAS mutant cell lines when compared to wild-type and BRAF V600E cells (Supplementary Fig. 8A-B; Supplementary table 8A-B). Interestingly, immune genes mutually exclusive to KRAS (e.g. BCHE, CCSER1, DDX60L) were also significantly differentially expressed in the transcriptome of cell lines with different KRAS mutations as compared to BRAF V600E mutants (Supplementary Fig. 8C; Supplementary table 8C). A subset of immune genes was expressed in a mutation specific manner, such as B2M (KRAS G12C), IFIT3 (G12D) and IL4R (G13D) in the IFNα pathway, IFIT3 (G12D), PARP14 (G12C), PTGS2 (G12V) in the IFNγ pathway, and ENPP6 (G12V), ARSD and IRF2BPL (G13D) genes with genomic deletions mutually exclusive to KRAS mutations were differentially expressed as compared to either BRAF V600E mutant or BRAF WT (Supplementary Fig. 8A-C; Supplementary table 8A-C). Moreover, some of the immune related genes such as IFI27, IFI35 (IFNα pathway), IFITM2, OGFR (IFNγ pathway) and CCSER1 (mutually exclusive to KRAS) were found to be differentially regulated in both RKO KRAS G13D vs RKO as well as HCT116 BRAF V600E vs HCT116 comparisons (Supplementary Fig. 8A-C; Supplementary table 8A-C). Taken together, 70% of IFNα pathway genes, 63% of IFNγ pathway genes, and 50% of KRAS mutually exclusive genes were significantly differentially expressed in the KRAS mutant cell lines (p ≤ 2.94E-12, hypergeometric distribution) [45–47].
Altered cellular metabolism as a consequence of Ras pathway mutations.
To understand how different KRAS mutations or the BRAF V600E mutation alter cellular metabolism, we performed UPLC-MS based metabolomic analysis. Whereas the PCAs comparing biological replicates demonstrated that the G12C cell clones exhibited separation among them, no significant differences between the two independent clones with KRAS G12D, G12V or G13D mutations were observed (Supplementary Fig. 9A-D). Pairwise comparisons revealed much fewer significantly altered metabolites and pathways (0–3), confirming that the independent clones were near identical in their metabolism. However, significant differences were observed between the mutant clones and their isogenic controls (Fig. 1C, Supplementary Fig. 9E). Altogether, the clear separation of Ras pathway mutant and control cells into discrete clusters in the PCA analysis was a strong indication that KRAS and BRAF mutations alter the metabolome. In a metabolic pathway analysis, where an average of 413 significant features were assigned to compounds in positive mode, and 303 in negative mode with FDR < 0.01, altered amino acid metabolism was observed following Ras pathway activation (Fig. 5A-D, Supplementary Fig. 10A-B and Supplementary table 9A-J). Across all mutants, we observed significant alterations in the tyrosine, histidine, arginine and proline metabolic pathways relative to their respective isogenic controls (Fig. 5A-D, Supplementary Fig. 10A-B and Supplementary table 9A-J) in agreement with previous studies [8, 48, 49]. Furthermore, certain amino acid metabolic pathways were significantly altered in a mutation specific manner, such as lysine metabolism in KRAS G12C and G12V (Fig. 5B and Supplementary Fig. 10A-B; Supplementary table 9C-D and 9I-J) and fatty acid oxidation in KRAS G12C and G13D in comparison to their respective isogenic controls (Fig. 5A and D; Supplementary table 9A-B and 9G-H). Additionally, the urea cycle/amino group, purine, pyrimidine and pentose-phosphate metabolic pathways were significantly altered by KRAS or BRAF mutation (Fig. 5A-D and Supplementary Fig. 10A-B; Supplementary table 9A-H). Interestingly, genes related to amino acid metabolism and fatty acid metabolism extracted from Kyoto Encyclopedia of Genes and Genomes (KEGG) database were significantly enriched in the transcriptome data (Supplementary Fig. 11A-J; Supplementary table 10A-J). Key metabolic pathway genes such as GOT1, which has roles in more than one metabolic pathway (i.e., tyrosine, histidine and proline metabolism) were differentially expressed in all KRAS mutants (Supplementary Fig. 11A, C and E). Taken together, the KRAS and BRAF mutations led to similar metabolic consequences, primarily affecting pathways connected to amino acids metabolism and the urea cycle/amino group metabolisms.
Carnitine metabolism is regulated by KRAS and BRAF mutations.
In the metabolomics analysis, significant alterations of L-carnitine derivatives were observed (Fig. 5E; supplementary Fig. 12). L-Carnitine is acylated by fatty acids in the cytosol and then transported into the mitochondria, where the fatty acid is released for β-oxidation. Carnitine is transferred back to the cytosol after release of fatty acids. Hence, carnitine plays an important role in fatty acid oxidation and mitochondrial metabolism [50]. Here, we observed higher abundance of free carnitine in KRAS mutant RKO cells, while its esters such as acetyl- and butyryl-carnitine were less abundant (Fig. 5E). Interestingly, the majority of all detected acylated carnitines were decreased in KRAS and BRAF mutant clones whereas medium-chain and long-chain carnitines such as tetradecanoyl, docanoyl and octanoyl carnitine were more abundant (Supplementary Fig. 12). Furthermore, differential expression of carnitine biosynthesis related genes such as CPT1 and 2 was evident in KRAS mutants compared to BRAF V600E (Supplementary table 2A, C and E). Members of the SLC22 transporter family, such as SLC22A5, play important roles in carnitine transport [50, 51] and mutation in SLC22A5 has been implicated in systemic carnitine deficiency related cardiomyopathy, skeletal myopathy and metabolic abnormalities [52]. Another member, SLC22A4, has been implicated in carnitine transport [51] and was differentially expressed in KRAS G12D/V versus wild-type cells (Supplementary table 2C and E). Notably, several uncharacterized members of this transporter family were differentially expressed here (SLC22A15 in KRAS G12D/V vs BRAF WT and KRAS G13D vs BRAF V600E (Supplementary table 2D, F and G), SLC22A17 in HCT116 BRAF V600E vs HCT116 (Supplementary table 2I), SLC22A18 in RKO KRAS G12C/D/V vs RKO BRAF WT (Supplementary table 2B, D and F) and SLC22A23 in RKO KRAS G12C/D/V/13D vs RKO (Supplementary table 2A, C, E and G) [51]. Thus, levels of carnitines and their biosynthetic genes were differentially regulated by mutant KRAS and BRAF.
{kind=link}