2. Biological studies
2.1. Antiproliferative activity
The antiproliferative activity of the synthesised compounds was tested using an MTS assay against the human colon carcinoma cell lines that had a wild-type (HCT116 p53+/+) and p53 negative status (HCT116 p53−/−). Additionally, the compounds were also tested for their cytotoxicity against normal cells – human dermal fibroblasts (NHDF). The results concerning the antiproliferative activity assays are shown in Tables 1 and 2.
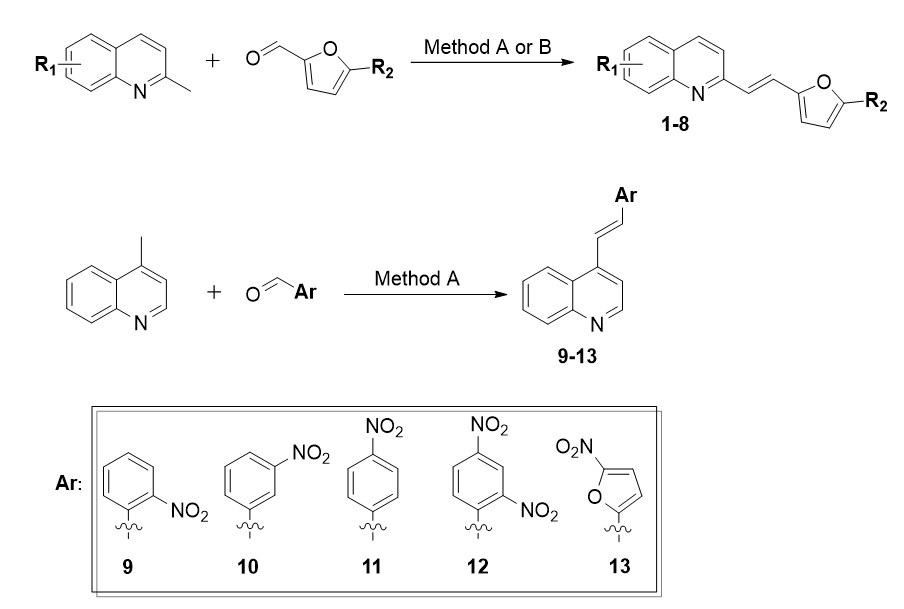
Based on our previous studies [3], and after performing cytotoxicity studies, it was concluded that both the replacement of phenyl with a furan ring in the 8-hydroxyquinoline derivative (1) as well as the further substitution of a methyl group to the furan ring at position 5 (2) rendered these compounds inactive towards the tested cancer cells (IC50 > 25 µM). However, the substitution of the furan ring with a nitro group in the same position 5 (3) significantly affected the activity of the tested derivatives and the IC50 values against both HCT 116 cell lines were below 0.2 µM. Cytotoxicity studies for this compound had previously been performed and our results were similar to those that had been obtained on human cancer cell lines of other origins [4, 5]. Moreover, the activity of derivative 3 was significantly higher compared to its analogue with a phenyl ring and NO2 group at position 2 (derivative 9b [3]) for which the IC50 values for HCT 116 p53+/+ and HCT 116 p53−/− were 11.72 µM and 2.61 µM, respectively (Fig. 2). Although the currently tested compound 3 was also cytotoxic against fibroblasts, it had a better selectivity index for HCT 116 p53+/+ than the previously tested compound with a phenyl ring. Acetylation of the hydroxyl group in the quinoline ring (4) did not improve the activity as was the case with the phenyl ring derivatives. Interestingly, in the case of the styrylquinolines, acetylation of the 8-hydroxyl group (derivative 14a [3]) improved the activity of the derivative against HCT 116 p53+/+ almost three-fold, although at the same time, it had a two-fold decreased activity against the HCT 116 p53−/− cells compared to its hydroxyl analogue 9b. Such large differences in the activity for lines that differed in their p53 status prompted us to further study the mechanism of action of this group of compounds. Therefore, in this research, we extended the investigations to the lines in which p53 mutations or deletions occur in order to understand the role of this protein in the context of the action of these derivatives.
The previously tested and published 4-hydroxyquinoline derivatives (including those with a nitro-substituted phenyl ring) did not exhibit any anticancer properties. In contrast, the 4-hydroxyquinoline derivative with a 5-nitrofuran ring (5) that was tested here exhibited significant antiproliferative features against cancer cells. Therefore, in this case, the change of heterocyclic ring with the analysed substituent also greatly improved the activity of that analogue. However, this compound was also the most toxic against fibroblasts (IC50 = 0.45 µM). On the other hand, adding the chlorine atoms in the 8-hydroxyquinoline ring at positions 5 and 7 resulted in a significant decrease in the IC50 values of 6 compared to 1 (from > 25 µM to 4 µM for HCT 116 p53+/+ and 2.7 µM for HCT 116 p53−/−), while still maintaining a selectivity against NHDF. Furthermore, the substitution of a nitro group onto the furan ring (7) increased the activity towards HCT 116 p53−/− two-fold. Interestingly, the activity of the acetoxy analogue 8 of the 7 derivative did not change much against cells with a deletion of p53; in contrast, it decreased against the cells with the wild-type p53. Interestingly, differences in IC50 values of 5,7-dichloro derivatives were observed between the tested cancer cell lines. All three were twice as active towards HCT 116 p53−/− than towards HCT 116 p53+/+. Preliminarily, these results suggested that the p53 protein was involved in the mechanism of action of these compounds. Furthermore, the derivatives of 5,7-dichloroquinoline 6–8 were considerably less active against cancer cells but had much better selectivity than their analogues without chlorine atoms 3–5. Moreover, it should be noted that in contrast to derivatives 3–5 the replacement of the phenyl ring with a furan ring did not increase the activity of 5,7-dichloro-8-hydroxyquinoline derivatives 7–8.
After a series of encouraging results, we decided to substitute position 4 instead of 2 in the quinoline ring, thus obtaining 4-styrylquinolines. We did not find any reports in the literature on the anticancer activity for 4-styrylquinoline derivatives, so we decided to investigate this. In the styryl moiety, we placed a phenyl ring substituted with a nitro group at various positions (9–12). Additionally, we have synthesised the 4-furanylvinylquinoline derivative with a furan substituent with a nitro group at position 5 (13). This compound was presented in paper [17], however, in the context of its antimicrobial properties, while its anticancer properties have not been previously described. After testing the cytotoxicity of these derivatives on the HCT 116 cells with the wild-type p53 protein and with a deletion of this protein (Table 2), the IC50 values in the case of 4-styrylquinoline derivatives 9–12 were more than 25 µM.
However, the antiproliferative activity of derivative 13 with furan a ring was surprising. The IC50 values for the HCT 116 cells were below 0.1 µM; therefore, we extended the investigation to a broader panel of cancer cell lines that diverged in their p53 status (Table 3). For this purpose, we selected two pancreatic cancer cell lines: AsPC-1, which is characterised by a deletion of the p53 protein [18] and PANC-1, which carries an R273H mutation [19]. This mutation also occurred in the glioblastoma U-251 cell line [20, 21]. An additional non-adherent line that carries the TP53 mutation that results in the absence of p53 is the K562 leukemic cell line [22]. Further studies confirmed the high activity of derivative 13 for which the IC50 values were maintained at a level below 0.1 µM for glioblastoma (U-251) and leukaemia (K562) and did not exceed 0.2 µM for the pancreatic cell lines. Moreover, 13 was selective towards fibroblasts, which was indicated by the SI values (3.35–9.41). Among all of the tested cell lines, the highest activity of the compound was found against the HCT 116 cells with the wild-type p53 protein (0.069 µM). However, in the case of both HCT 116 cell lines, 13 was more than four times more active than the commercially available drug, Doxorubicin, while at the same time it was also more than four-fold less toxic against fibroblasts. The p53 protein also interacts with the metabolic pathways that are associated with glucose transport [23] as well as small molecules [24]. In light of the above-mentioned facts, we also determined the cytotoxicity of the GLUTs inhibitor – BAY-876, which is based on a quinoline scaffold. Bay-876 was the most active in the PANC-1 cells with a R273H mutation of p53. Interestingly, for the cell line with the same mutation (U-251), it was not active at 25 µM. In HCT 116 p53+/+, the IC50 value was 1.71 µM while for this same line with a deletion of the TP53 gene, the activity of this inhibitor decreased more than six-fold. Furthermore, BAY-876 was also toxic to normal cells.
To confirm the cytotoxicity rank, the viability of the cells was also estimated on a 3D spheroid model, which better represented a solid tumour. In contrast to the results that were obtained on the monolayer model, the tested derivative was the most active against the U-251 spheroids (IC50 = 0.34 µM). However, derivative 13 was also characterised by a high activity against the colon cancers spheroids for which the IC50 values were approximately 1 µM. The cell lines that represented pancreatic cancer were less responsive to derivative 13, which was similar to the 2D model. It is well known that pancreatic cancer exhibits a high drug resistance [25, 26]. However, the activity of derivative 13 was confirmed on the 3D model that reflected a real tumour, which is undoubtedly a positive result and a reason for further, more in-depth studies to clarify the mechanism of action of 4-furanylvinylquinoline derivative 13.
2.2. Cell cycle inhibition
When analysing the antiproliferative activity of the tested compounds, we decided to determine the molecular mechanism of action of the most promising derivative 13 in three cell lines with different statuses of p53 for which the derivative has the highest activity. In order to achieve this goal, we started by studying the changes in the cell cycle in order to determine why proliferation stops. To evaluate the effect of derivative 13 on the cell cycle progression, we used flow cytometry. After a 24-hour treatment, we observed a significant inhibition in the G2/M phase in both HCT 116 cell lines (Fig. 3A). The population of cells in this phase increased from 30.81–49.78% and from 34.35–44.95% in HCT 116 p53+/+ and HCT 116 p53−/− cells compared to control cells, respectively. On the other hand, in the case of the U-251 cells, the compound inhibited the cell cycle in the S phase. There was a pronounced increase in the percentage of cells from 20.44–35.59%. The representative histograms from the flow cytometry are shown in Fig. S1 in SI.
A. The influence of derivative 13 (concentration 2xIC50) on the cell cycle progression measured by flow cytometry in the HCT 116 p53+/+, HCT 116 p53−/− and U-251 cell lines after a 24-h incubation. The results are presented as the mean ± SD and summarised in the chart. The data were analysed using an unpaired t-test with Welch’s correction: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to the untreated cells (control).
B. The charts present the relative mRNA expression of CDK1 in the tested cell lines after 24- and 48-h treatment with derivative 13 at various concentrations. The statistical analysis was performed using a one-way ANOVA with Bonferroni’s post-hoc test: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to the untreated cells (control).
In the next step we decided to investigate the impact of derivative 13 on the mRNA level of CDK1 – the gene encoding one of the cell cycle regulation proteins at various concentrations and time points (Fig. 3B). In the HCT 116 p53+/+ cells after the application 0.5 µM of derivative 13, we noticed a slight increase in the expression of CDK1 after both 24 and 48 hours. Interestingly, after the 24-hour treatment with 1 µM, we did not observe any significant changes relative to the untreated cell, but after 48-hours of incubation, the mRNA level rapidly increased. In contrast, after 24h, the level of CDK1 was two-fold higher in the HCT 116 cells with a deletion of TP53 than in the control cells at a concentration of 0.5 µM and around one and the half times in the 1 µM samples and considerably increased after 48 h. We also observed more than a two-fold upregulation of the CDK1 expression in the U-251 cells after 24h. However, after a further incubation time, the mRNA level of this gene was similar to that in the untreated cells.
To determine whether and to what extent the compound interacts with DNA, we also performed a spectrophotometric study on CT-DNA. We observed a moderate hypochromism of derivative 13 (10.97%). (Fig. S2). Nevertheless, under the effect of stress-induced DNA damage in cells, CDC25C as well as p53 were activated, which inhibited the CDK1 and cyclin B1 complex. This had the effect of arresting the cell cycle in the G2/M phase in an attempt to repair the DNA damages or induce apoptosis [27, 28]. Abnormal CDK1 activation is often thought to be connected with tumourigenesis. However, in recent years, there have been reports that CDK1 is also involved in the induction of cell death via apoptosis [29, 30].
2.3. Apoptosis induction
Because derivative 13 affected the cell cycle progression, we decided to determine the type of induced cell death focusing on the apoptosis pathway. The analysis was performed by measuring the green fluorescence of Annexin V – FITC dye conjugate using capillary flow cytometry. Positive staining is observed when the annexin V binds to the disintegrated membrane of apoptotic cells. Generally, after treatment with derivative 13, there was a strong increase in the percentage of apoptotic cells in all of the cell lines that were tested, namely from nearly 6–40% and 30% for HCT 116 p53+/+ and HCT 116 p53−/− cells, respectively (Fig. 4A). However, the highest percentage of apoptotic cells was observed in the U-251 cells where their fraction had changed from approximately 13% in the control to 66% in the treated cells. The representative histograms from the flow cytometry are shown in Figure S3 in SI.
A. The effect of derivative 13 (concentration 0.5 µM) on apoptosis induction in the HCT 116 p53+/+, HCT 116 p53−/− and U-251 cell lines after a 48-h incubation. The percentage of live, apoptotic (early and late), and dead cells from all of the performed experiments for each of the cell lines are shown in the chart as the mean ± SD. A statistical analysis of data was performed using an unpaired t-test with Welch’s correction: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to the untreated cells (control).
B. The charts present the relative mRNA expression of TRAIL, DR5 and caspase-8 in the three tested cell lines after treatment with derivative 13. Statistical analysis was performed using a one-way ANOVA with Bonferroni’s post-hoc test: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to the untreated cells (control).
After analysing the obtained results, we hypothesised that the compound affects p53, and therefore we decided to investigate some of the targets that are associated with extrinsic apoptosis. Because certain chemotherapeutic agents can induce the tumour necrosis factor (TNF), which is related to the apoptosis-inducing ligand (TRAIL)-mediated apoptosis [31], we decided to investigate the effect of derivative 13 on the expression of TRAIL, TRAIL receptor 2 (TRAILR2/DR5) and the extrinsic pathway related to caspase 8 (Fig. 4B). In all of the cases, there was a decrease of TRAIL mRNA level; however, in the HCT 116 p53−/− cells, it was not significant. Certain cytostatic compounds such as etoposide, which induce apoptosis by activating caspase 8, did not induce an mRNA expression of the death receptor ligands [32]. The compound that was tested here clearly increased the expression of DR5 after just 24 hours of treatment in the U-251 cells and the increase was observed in HCT 116 p53−/− to a lesser extent after that time. In contrast, in the colorectal cancer cells with the wild-type p53, there was an increase in the expression of the gene encoding this receptor after 48 hours. According to the results that were obtained by Wachter et al., both the wild-type and mutated p53 in cancer cells play important roles in the induction of the extrinsic apoptosis pathway [33]. Our research also confirmed this correlation. However, the authors emphasised that the p53 status had no significant effect on the changes in the expression of apoptotic targets that are regulated by p53 including DR5. In the results presented here, after incubation with derivative 13, we observed different levels of the increase in the DR5 expression depending on the cell line. P53-mediated apoptosis via enhancing the expression of DR5 was observed in HCT 116 with the wild-type p53 after treatment with 5-fluorouracil [34] as well as with prostaglandin A2 (PGA2) [35]. Moreover, we observed a greater sensitivity to the induction of apoptosis in HCT 116 p53+/+ than in HCT 116 p53−/−. Similar observations were recently made by Willms and colleagues after treatment with clinically tested agonistic antibodies against the TRAIL receptors, which indicates an important role of p53 [36]. Nevertheless, our results showed the highest sensitivity to the induction of apoptosis in the p53-mutant cells, which was also confirmed by the high expression level of DR5 after 24h. After this time, we also observed a significant up-regulation of caspase-8, which is one of the major molecular targets in executing the extrinsic apoptotic pathway, in the U-251 cells [37]. A less pronounced change in the mRNA level of this gene was observed in both HCT 116 cell lines. This might be related to a recent report that showed that cancer cells can use caspase 8 to overcome the checkpoint between the G2/M phases of the cell cycle. The results presented above indicated that cell cycle arrest occurred in this phase in HCT 116 cells. Interestingly, p53 might also act as a key regulator of the caspase-8 expression [38]. Liu and colleagues reported that an increase in the mRNA level expression of caspase-8 was not observed after treatment with a cytostatic compound in cancer cells that lacked p53. We made a similar observation in the p53-deficient HCT 116 cells. Our results also confirmed that the tested derivative induced the expression of the selected targets of the extrinsic apoptosis based on the p53 status in cells. Furthermore, it has also been reported that a higher level of DR5 is not only associated with p53 activation but also with the DNA damage that is caused by ROS [39]. Therefore, in the next step, we decided to determine the impact of derivative 13 on oxidative stress.
2.4. Oxidative stress induction
To determine the ROS level the fluorescent CellROX® Green Reagent measured by spectroscopic multi-plate reader was used. A kinetic increase of the ROS level was observed in each of the tested lines within 24 h (Fig. 5A). The most noticeable increase was observed in HCT 116 with a p53 deletion. The ROS level was several percent higher compared to the control after only 3h. After 24h the amount of ROS increased by as much as 50% relative to the untreated cells. Similar observations were made in the case of the HCT 116 p53+/+ cells. However, the generation of ROS was less pronounced, especially at the first time points. Nevertheless, after 24h, the level of ROS was about 40% higher. The least visible increase of ROS was observed in the U-251 cells. The highest (20% growth) occurred after 9 h and 24 h. Interestingly, after a 12-hour incubation, there was a slight decrease. The intracellular ROS levels are rapidly regulated by activated p53 by initially decreasing and then subsequently increasing them [40]. A weaker response of the ROS generation in these cells might have been caused by the fact that U-251 cells are resistant to exposure to oxidative stress. Liu et al. demonstrated that a high concentration (300 µM) of hydrogen peroxide (H2O2) inhibited the proliferation of U-251 cells after 24 h [41]. Additionally, cancer cells with a p53 mutation have a higher level of ROS than those with the wild type. Some p53 mutants can gain oncogenic features that are described as “gain-of-function” (GOF). Among them there are U-251 cells [20]. By controlling the signalling pathways that are associated with the redox balance, the GOF p53 mutants promote an elevated basal level of ROS in cancer cells [42, 43]. Therefore, in the case of the tested glioblastoma cells, derivative 13 had a smaller effect on ROS generation than it did in the colon carcinoma cells.
A. The line charts present the effect of the tested compound on the generation of ROS. The results are presented as the mean ± SD of three independent experiments. The data were normalised to the untreated cells (control) and analysed using the unpaired t-test with Welch’s correction: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
B. The column charts show the changes in the relative mRNA expression (presented as the mean ± SD) of CAT and MnSOD after 24- and 48-hour treatment with derivative 13. The results from three independent experiments are presented. The statistical analysis was performed using a one-way ANOVA with Bonferroni’s post-hoc test: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to the control.
The overproduction of ROS might involve the induction of apoptosis [3] and might also have an impact on the regulation of the expression of some of the enzymes that are involved in maintaining the appropriate redox balance. Among the most important ones are manganese superoxide dismutase (MnSOD) and catalase (CAT) which were examined by qRT-PCR. MnSOD as one of the antioxidant regulators that are located in the mitochondrial matrix, which maintain the appropriate level of ROS by converting superoxide anion (O2.−) into hydrogen peroxide (H2O2), which is next dismutated into water and oxygen by CAT in the cytoplasm [44, 45]. After treatment with derivative 13, we observed an increase in the MnSOD expression at both time points and in all of the tested cell lines (Fig. 5B). These results confirmed that the antioxidant system was activated in the cells due to disturbances in the ROS homeostasis. However the mRNA level of MnSOD was particularly marked in the cells with p53 (HCT 116 p53+/+ and U-251). As was reported by Zhao et al., a compound that activates p53 might also induce the translocation of this protein into the mitochondrial matrix and then interact with MnSOD [46]. The regulation of the MnSOD expression by p53 can proceed in two ways. Some studies have shown that p53 inhibits the expression of this antioxidant enzyme [47]. On the other hand, other results have shown that p53 might enhance the expression of MnSOD through an interaction with the other transcription factors that are associated with cell proliferation such as NF-κB [48]. The overexpression of MnSOD by the activation of p53 was observed in human lymphoblasts [49]. It caused an increase in oxidative stress, which caused apoptosis. In contrast to MnSOD, the CAT levels were lower than in controls. Given that the levels of these two genes were recorded at the same time points, the results demonstrate that the antioxidant system of the cells was activated. Because the stronger radicals (such as HO. or O2.−) are inactivated first, followed by the weaker ones such as H2O2, the expression of the ROS scavengers may vary over time. The results obtained in our study confirm the hypothesis that the activation of p53 might upregulate the gene expression of MnSOD because in HCT 116 p53−/−, the MnSOD expression was much less noticeable than in the case of the other lines with p53. In our research, we also observed an increase in MnSOD and apoptotic cell death. Derivative 13 induced oxidative stress via the increased production of intracellular ROS and affected the activation of the antioxidant system.
2.5. Protein level analysis
To determine the effect of derivative 13 on the cellular pathways and protein expression Western Blot analysis was performed. The activation of the p53 protein was observed after a 24-hour incubation in cells with the wild type (HCT 116 p53+/+) (Fig. 6A) and mutation (U-251) at both time points (Fig. 6C). In the glioma cells, higher relative levels of p53 were observed after 48 hours. As might be expected, in the HCT 116 p53−/− cell line, we did not observe its occurrence due to the deletion of the gene encoding this protein. The first important conclusion of the analysis is undoubtedly the involvement of the p53 protein in the mechanism of action of derivative 13. The HIF-1α protein was also analysed, but its activation was not found in any of the tested cell lines (data not shown). Therefore, the involvement of hypoxia in the mechanism that affected the antiproliferative activity of derivative 13 was excluded.
The results obtained from the analysis of the generation of reactive oxygen species led to analyse the level of the HO-1 protein – an enzyme that is responsible for maintaining normal cellular homeostasis and that also plays an important role in the response to oxidative stress. A concentration-dependent increase of HO-1 was observed in all of the cell lines (Fig. 6). The highest level was observed at 1 µM after 24h (more than twelve-fold in HCT 116 p53+/+, six-fold in HCT 116 p53−/− and four-fold in U-251) (Fig. S4). Interestingly, an increase in HO-1 after treatment with doxorubicin was only observed in the HCT 116 p53+/+ cells, whereas in the other two lines, there was a decrease in this enzyme. Furthermore, a decrease in HO-1 relative to the control was observed in HCT116 cells with p53 deletion after 48h in contrast to the wild-type cells where levels were still higher than in the control. Derivative 13 induced oxidative stress via the increased production of intracellular ROS, which affected the activation of the antioxidant system. However, the redox disturbances were so strong that the activation of the scavengers could not cope with the excessive stress and the cells switched to the cell death pathway, more specifically apoptosis. Recent studies on human osteosarcoma U2OS cells have also shown that HO-1 induction was associated with cell cycle arrest in the G2/M phase [50]. The results presented in this paper also showed significant cell cycle inhibition in this phase in the cells of both of the studied colorectal cancer lines. Therefore, the increased activation of HO-1 in the HCT 116 p53+/+ and p53−/− cells might have been due to its involvement in the inhibition of the cell cycle progression. The U-251 line exhibited the lowest relative increase in the HO-1 levels compared to the control cells.
In the present study, at both concentrations, derivative 13 caused a more than two-fold increase in the p21 protein expression in the HCT 116 p53+/+ cell line, which persisted after 24 and 48 hours. In the case of the HCT 116 p53−/− cells, a concentration of 0.5 µM stimulated the activation of this protein after a 24-hour incubation, while a concentration of 1 µM had an inhibitory effect on the p21 expression. This results indicate that the p21 protein activation was independent of the p53 protein as was described in [51]. The overexpression of p21 also causes cell cycle inhibition via its binding to the CDK1/cyclin E complex [52]. In addition, it was proven that the inhibition of the cell cycle in the G2/M phase that is caused by CP-31398 styrylquinazoline was associated with the p21 activation [53]. In the tested colorectal cancer cell lines, derivative 13 clearly induced cell cycle arrest precisely at this phase, and therefore the higher levels of p21 were most likely due to its involvement in the cell cycle regulation rather than apoptosis induction. The p21 protein does not accumulate when the cycle is inhibited in the S-phase even when cells are treated with DNA-damaging agents that cause it to accumulate to a degree that is easily detectable by the Western Blot method [54]. The cell cycle in the U-251 cell line was inhibited precisely in the S-phase, hence, it is likely that the p21 protein expression was not detected in the glioma cells tested (data not shown).
The results that were obtained for the CDK1 protein overlapped with those for the cell cycle analysis using flow cytometry (Fig. 3A) and the expression analysis of the gene encoding this kinase (Fig. 3B). In the HCT 116 p53+/+ line cells, a slight increase in the expression of this protein was generally observed at both 24 hours and 48 after the treatment with derivative 13 (Fig. 6A). The level of the transcribed protein was quite similar to the CDK1 gene expression. However, there was a lack of this correlation after 48 hours with 1µM of the tested compound. The CDK1 gene expression at this concentration increased sharply, while the protein levels decreased significantly relative to the untreated cells. The most likely explanation for this could be the activation of various metabolic processes in order to protect the cells from the oxidative stress that was induced by the administration of the compound. Therefore, there was such a sharp increase in the expression of the gene encoding this kinase that as many cells as possible managed to undergo mitotic division. However, a full translation did not occur through the damage to the mechanism of protein synthesis such as at the level of ribosomes, which in turn is the result of excessive levels of ROS [55]. This resulted in a decrease in the CDK1 levels and cell cycle arrest in the G2/M phase. On the other hand, after a 24-hour incubation with derivative 13 at both tested concentrations, the level of the CDK1 protein in the HCT 116 p53−/− cell line was quite similar to that in the control cells. After 48 hours, an intense activation of CDK1 was detected, which in turn coincided with the results that were obtained at the mRNA level. The observed phenomenon was probably related to the downward trend of the p21 protein at this time, which had an inhibitory effect on CDK1 kinase. Interestingly, the sharp increase in CDK1 levels in this cell line after 48 hours was also observed after the doxorubicin treatment. In the U-251 glioma cells, there were no significant differences in the protein levels between the untreated cells and those that were incubated with derivative 13. The only significant accumulation of this protein occurred after 48 hours at the higher tested concentration. These results suggest that the inhibition of the cycle occurred in the S phase.
The results of the analysis of induced cell death prompted us to determine the levels of the proteins that are associated with apoptosis induction. The tested compound affected the activation of Fas protein in the colorectal cancer cells. In the HCT 116 p53+/+ cell line, a more than three-fold increase in the level of this marker of the extrinsic apoptosis pathway was observed after 24 hours (Fig. S4A). The intensity of the Fas expression in this cell line was also maintained after 48 hours but to a lesser extent. In contrast, in the HCT 116 p53−/− cells, the increase in the Fas protein expression occurred after 48 hours and was four times higher than in the untreated cells (Fig. S4B). In the case of both of the tested colon cancer lines, there was no significant increase in the level of this protein after the doxorubicin treatment. The results that were obtained corroborated those that were obtained from the mRNA expression studies of the other selected markers of the extrinsic apoptosis pathway, which indicated this cell death pathway in both of the studied HCT 116 lines. The difference in the activation time could have been related to the presence of the p53 protein in the cells. There are reports on the regulation of the apoptosis pathway in which Fas participates depending on its p53 status, which only involves the Fas-specific binding to the wild-type p53 protein [56]. It is also known that the transcription of the gene encoding Fas can be regulated by the nuclear factor NF-κB [57], Wig-1 [58], or p73 [59], which could be related to the later activation of this protein in the HCT 116 p53−/− cells. On the other hand, it has been proven that Fas is not always expressed in the cells that express the wild-type p53, instead it was detectable in cells with a homozygous TP53 deletion [60]. Given the above information, it was not possible to indicate to what extent the p53 protein regulated the Fas expression. Nevertheless, derivative 13 undoubtedly affected the upregulation of this membrane protein. Interestingly, the level of the Fas protein was not detected in the U-251 cells by the Western Blot method (not shown). In this case, it cannot be excluded that mutated p53 played an important role in its repression. Notably, Zalcenstein et al. showed that the mutant p53 protein inhibited the expression of the gene encoding the Fas receptor protein, and the degree of this inhibition varied depending on the type of the p53 mutation [61].
In contrast to both of the HCT 116 lines, the proteolysis of the PARP-1 protein was observed in the glioma cells after 48 hours. The level of the 89 kDa product, which confirmed apoptosis after treatment with the tested derivative 13 was higher than after the doxorubicin treatment (Fig. 6C). By analysing the previous results that prove the induction of apoptosis, PARP-1 could also be seen as marker of the DNA damage that affected the topoisomerases, which are enzymes that are essential during DNA replication. This is a fact that has been described for the glioma cell lines including U-251 [62]. It is therefore possible that the molecular substrate in the S-phase cell cycle inhibition after treatment with derivative 13 was precisely the interaction of the compound with the topoisomerases.
Considering the obtained results (the influence of the studied derivative on p53 activation) and the fact that wild-type p53 positively regulates oxidative phosphorylation and inhibits glucose metabolism through the inhibition of glucose transporters, among others, GLUT-1 and GLUT-4 we decided to performed further analysis. In turn, a p53 mutation increased the expression of these glucose transporters, which favours aerobic glycolysis over oxidative phosphorylation, i.e., p53 mutations contribute to the Warburg effect.
Therefore, we decided to test the effect of the tested compound on the level of the most popular glucose transporter GLUT-1. In the HCT 116 p53+/+ cells, a 24-hour incubation with derivative 13 resulted in a decrease in the level of this glucose transporter, whereas a rapid activation of this protein was observed after 48 hours. The results obtained for this cell line evidenced the aforementioned mechanism of the regulation of the glucose transporter activity by the wild-type p53 protein. Indeed, after 24 hours, the activation of this protein was observed in these cells, which coincided with the inhibition of GLUT-1. In contrast, after 48 hours, no activation of p53 was observed, which resulted in a sharp increase in the expression of the glucose transporter for cell defence. The validity of the described conclusions was confirmed by the results that were obtained for doxorubicin in which the p53 activation occurred at both tested time points with a concomitant decrease in the GLUT-1 levels. On the other hand, despite the slight increase in the GLUT-1 level after a day of incubation with 0.5 µM of derivative 13, a decrease in the level of this transporter was observed in the HCT 116 p53−/− cells, which would indicate a p53-independent GLUT-1 inhibition. Notably, in the U-251 glioma cells, the compound caused such a significant decrease in the level of this protein compared to that untreated cells that it was not detectable by the Western Blot method. The results that were obtained from the measurements of the expression of this protein were so interesting and at the same time inconclusive that we additionally decided to determine the effect of the tested derivative 13 on the expression of the genes encoding selected glucose transporters.
2.6. Expression of glucose transporters genes
To verify the effect of derivative 13 on glucose transporters at the mRNA level, the expression of the genes encoding the best-characterised glucose transporters in cells was quantified in real-time. In addition to GLUT-1, the effect of the tested derivative on changes in the expression of GLUT-3 and GLUT-4 was also evaluated (Fig. 7). Furthermore, the inhibitor BAY-876, whose structure is based on a quinoline backbone, was used as a positive control. The BAY-876 inhibits all glucose transporter proteins GLUT1-4 with preference to GLUT-1 (selectivity over 100 times) [63]. Inhibition of the GLUT proteins may induce hypoxia through metabolic changes, which in turn is the key factor of increasing expression and translocation of the proteins [64]. The results obtained from the analysis of the GLUT-1 expression in the HCT 116 p53+/+ cell line correlated with those that were obtained at the protein level. Interestingly, after 24 hours, the tested compound caused a greater inhibition of the GLUT-1 expression than BAY-876 in which an increase in the mRNA level of this gene was observed after this time. The opposite situation was observed after 48 hours after which the mRNA level of GLUT-1 increased after incubation with derivative 13, whereas it decreased after the application of BAY-876. In the HCT 116 p53−/− line cells, a significant decrease in GLUT-1 levels was observed after treatment with the furanylvinylquinoline derivative at both time points that were
analysed, whereas the BAY-876 treatment increased its expression. In contrast, in the U-251 glioma line, in which there was a strong inhibition of the GLUT-1 protein, the test compound itself did not significantly affect the expression of the gene encoding this transporter nor did BAY-876.
Derivative 13 also did not significantly affect the changes in the GLUT-3 expression in both of the tested HCT 116 lines. In both the wild-type p53 and TP53 knockout cells, a 24-hour incubation with the tested derivative at a concentration of 0.5 µM resulted in a slight decrease in the GLUT-3 mRNA levels, while the higher concentration caused a slight increase in the expression of the gene encoding this transporter. The observed effect persisted after 48 hours as well. The application of BAY-876 caused an increase in GLUT-3 expression after 24 hours. However, after 48 hours, a significant decrease in the mRNA levels was observed in the HCT 116 p53+/+ cells, whereas in the HCT 116 p53−/− cells, there was a strong increase in the expression of this gene. In the case of the U-251 line, after 24 hours, both derivative 13 and the reference BAY-876 induced the upregulation of the GLUT-3 mRNA levels, whereas after 48 hours, a decreasing trend was observed.
After analysing the results for GLUT-4, it was concluded that when the derivative 13 was tested in the HCT 116 p53+/+ cells, it did not play a significant role in regulating the expression of this gene. No significant changes were observed in contrast to the samples that were treated with BAY-876 in which there was a significant decrease in the GLUT-4 mRNA levels. More varied results were obtained in the HCT 116 p53−/− line, in which, after treatment with derivative 13 at a concentration of 0.5 µM, the effect was the same as after incubation with BAY-876. In contrast, the derivative when tested at a concentration of 1µM stimulated upregulation of GLUT-4 mRNA. In the U-251 glioma cells, derivative 13 at a higher concentration caused an increase in the mRNA level of this glucose transporter after 24 hours, while after 48 hours there was a clear inhibition of the GLUT-4 expression. However, the expression of this gene was most inhibited by BAY-876.
To summarise the results, it was concluded that an inhibitory effect on glycolysis that was dependent on the presence of the wild-type p53 protein was observed in the 116 p53+/+ HCT cells. According to the literature, p53 can inhibit glucose transport by directly reducing the expression of the genes encoding GLUT-1 and GLUT-4 [65] and also indirectly by inhibiting GLUT-3 transcription as a result of the inhibition of nuclear factor NF-κB [66]. In contrast, the observed effect in the HCT 116 p53−/− cells contradicted the involvement of the p53 protein in the inhibition of the expression of the glucose transporters, in particular GLUT-1. Similar conclusions were drawn after analysing the expression profiles of the genes that were studied in glioma cells with mutp53. In this case, one explanation is that p53 plays conflicting roles in glucose metabolism. On the one hand, it inhibits glycolysis, which prevents ATP production, while on the other hand, it promotes oxidative phosphorylation to maximise the production of the 32–34 molecules of ATP by utilising a small amount of pyruvate in the mitochondria. The p53 protein may balance the maintenance of glucose homeostasis with the generation of ROS, which is mediated by oxidative phosphorylation [23]. In contrast, tumour-associated mutp53 does not affect GLUT-1 expression but stimulates the Warburg effect by promoting the translocation of this glucose transporter to the plasma membrane. However, mutp53 has been shown to largely abrogate the stimulatory effect of the mutations on the Warburg effect in cells [67]. This fact underscores the fact that the results obtained in the U-251 line after the derivative 13 treatment are very promising for the future.
In addition, Zhao et al. observed that a dihydroxy derivative of flavone reduced glycolysis and the proliferation of cancer cells expressing the wild-type p53 protein but not mutp53 [68]. In contrast to this compound, derivative 13 downregulated the GLUT-1 expression and inhibited the growth of the U-251 line cells that were tested with mutp53. It is worth noting that the authors of the above studies ruled out the induction of apoptosis via the generation of ROS. On the other hand, in the analysis of the current results, the pronounced overproduction of intracellular ROS in all three cell lines should also be taken into account. According to the literature, a rapid stimulation of glucose uptake can occur at the level of the regulation of the intrinsic activity of the glucose transporters as well as their translocation, which is induced by ROS-induced signals. This is part of the adaptive mechanism that is activated by oxidative stress [14]. Increased ROS level by stabilization of HIF-1 may enforced the expression of GLUTs as well as further metabolic changes in cancer cells [69]. The inclusion of such a metabolic pathway could have contributed to the effects that were observed in the mRNA levels of the glucose transporters that were studied in a manner that was independent of the p53 protein status. Most likely, however, the inhibition of GLUTs, particularly GLUT-1, only complemented the molecular mechanism of the action of derivative 13 based on the generation of intracellular ROS, which resulted in the induction of the extrinsic apoptosis pathway. The observed effect is always a result of various factors, as evidenced in our studies comparing ROS secretion, differential MnSOD response, and the impact on GLUT. Therefore, additional studies correlating GLUT receptor expression with other metabolic pathways altered by compound 13 are necessary.
{kind=link}