While ensuring the effectiveness of medication in chronic disease is paramount, recognising the importance of managing both safety and cost through a holistic approach, as encapsulated by the medication triangle, contributes to optimal and enduring treatment outcomes. Within the context of T2DM management, TZDs serve as a prime example of how the medication triangle plays out in practice. Despite showcasing efficacy in maintaining glycaemic control and offering affordability, TZDs fall short in the medication triangle, primarily due to concerns surrounding their safety profile (Association, 2023). The emergence of clinical evidence demonstrating a link between TZD usage and HF has fundamentally reshaped the risk–benefit profile of these medications, leading to marked restrictions in their clinical use (Administration, 2010; 2012; De Flines & Scheen, 2007). Nevertheless, the exact mechanisms responsible for triggering or aggravating cardiac events in response to TZD usage are still unclear, impeding a holistic understanding of this complex interplay. Motivated by the obscurity surrounding the mechanistic nature of TZD cardiotoxicity, this study introduced a comprehensive multi-omics approach to unravel the hitherto undeciphered pathomechanisms driving this adverse effect.
4.1 Decoding the Metabolic Remodeling of AC16 Cardiomyocytes Following TZD Exposure
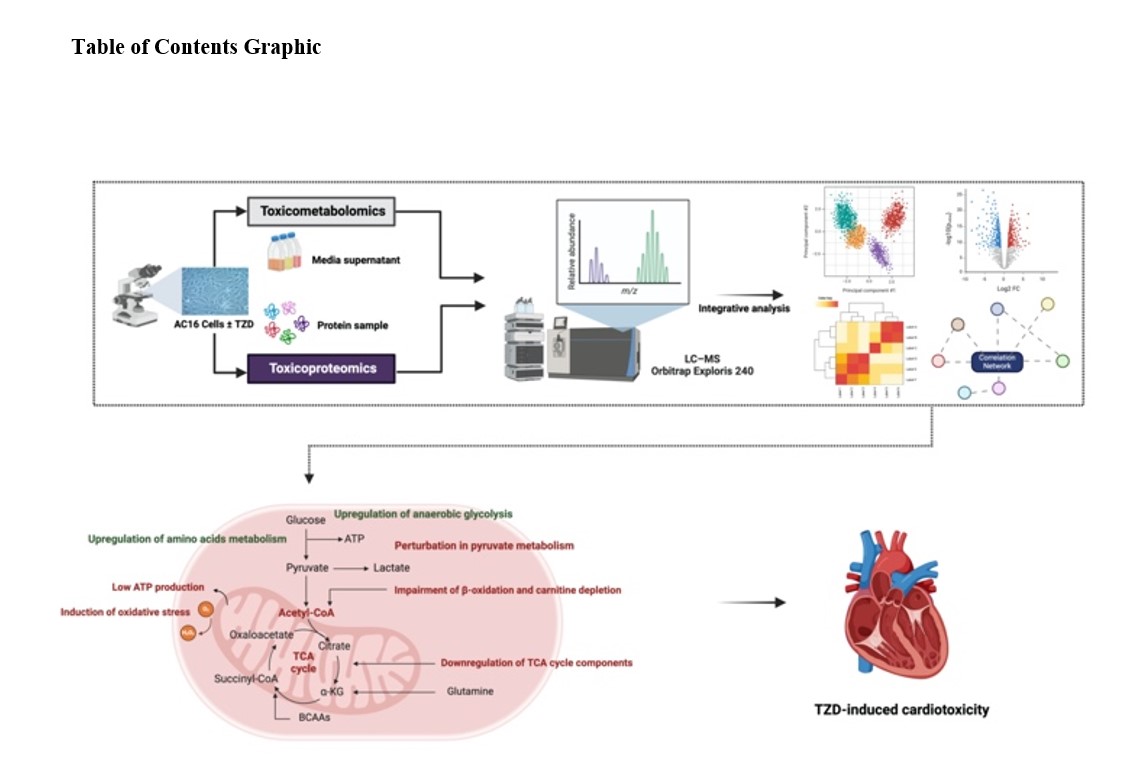
HF is demonstrably characterised by early disruptions in cardiac energy metabolism, preceding discernible structural alterations. Our multi-level molecular profiling corroborates this notion, uncovering distinct patterns of metabolic reprogramming across multiple pathways, culminating in perturbed cardiac energetics. Our analysis revealed a coordinated downregulation of crucial pathways involving oxidative phosphorylation (OXPHOS), the citric acid cycle (TCA), pyruvate metabolism, and fatty acid synthesis in response to TZD treatment. Conversely, increased activity was observed in glycolysis, the pentose phosphate pathway, and amino and purine metabolism. This coordinated pattern strongly suggests a marked switch in AC16 metabolic fate manifested as metabolic shift from fatty acid oxidation towards anaerobic glycolysis, potentially contributing to cardiotoxicity progression.
In the context of TCA and OXPHOS, a marked downregulation in mitochondrial NAD(P)+-dependent malic enzyme (m-NAD(P)-ME), a protein with a prominent role in catalysing the oxidative decarboxylation of malate to pyruvate, feeding into the TCA cycle (Hsieh et al, 2019) was noted following PGZ treatment. However, ROSI treatment induced substantial downregulation of fumarate hydratase (FH), a homotetrameric mitochondrial enzyme catalysing the reversible hydration of fumarate to malate within the TCA (Valcarcel-Jimenez & Frezza, 2023). This downregulation led to a marked accumulation of fumarate, mirroring its observed abundance in TZD-treated cells. Another crucial protein perturbed by ROSI treatment is malate dehydrogenase, a key enzyme in the oxidation of pyruvate and TCA and a member of the malate-aspartate shuttle (Ahn et al, 2020). This metabolic pathway functions as a conduit for electrons generated during glycolysis, facilitating their transfer from the cytosol to mitochondria for OXPHOS (Ahn et al, 2020). Malate dehydrogenase catalyses the reversible conversion of malate to oxaloacetate, enabling NADH transfer from the cytoplasm to mitochondria (Ahn et al, 2020). Therefore, given its established role, disruption of malate dehydrogenase by ROSI critically impairs the malate-aspartate shuttle, leading to reduced NADH transfer to mitochondria and compromising OXPHOS. Furthermore, ROSI treatment induced a striking enrichment in the nitrogen metabolism pathway. Notably, the glutamate dehydrogenases (GLUD1), key enzymes converting glutamate to α-ketoglutarate (α-KG), exhibited significant downregulation (Craze et al, 2019). This resulted in a marked accumulation of glutamate, as confirmed via our analysis, and compromised aerobic energy output, as α-KG serves as a crucial intermediate in the TCA cycle.
Essential for normal cardiac function, long-chain fatty acids serve as the preferred energy source for the heart, enabling efficient ATP production through mitochondrial β-oxidation while simultaneously contributing to the structural integrity and function of cellular membranes by replenishing their lipid composition (Yamamoto & Sano, 2022). Our complementary analysis revealed a compelling downregulation of the fatty acid synthesis pathway, evidenced by the marked decrease in acyl-CoA synthetase long chain family member 1 (ACSL1) expression in both PGZ- and ROSI-treated cells. This significant ACSL1 downregulation, a key enzyme responsible for long-chain fatty acid activation and β-oxidation initiation (Roelands et al, 2019), aligns with the observed reduction in cellular fatty acid levels, including palmitic and stearic acids, following TZD treatment, which together shed light on potential impairment in fatty acid oxidation that underlined the observed changes in cellular energy metabolism following PGZ and ROSI treatment. The present data support the observations of Shekar et al., who reported increased degradation of proteins essential for mitochondrial fatty acid metabolism resulting in deficits in fatty acid oxidation in a Sprague Dawley rat model of transverse aortic constriction-induced moderate HF (Shekar et al, 2014).
Beyond directly impacting fatty acid synthesis and β-oxidation, TZDs orchestrate a broader metabolic reprogramming reverberating through amino acid metabolism pathways markedly linked to fatty acid oxidation. This multifaceted effect, revealed by our integrated pathway analysis, manifests as a significant downregulation of lysine degradation in cardiac cells. This combined pathway analysis unveils an accumulation of lysine and its precursor, L-α-aminoadipate, coupled with a significant suppression of dihydrolipoamide dehydrogenase, a critical pyruvate dehydrogenase complex subunit vital for β-oxidation and pyruvate-to-acetyl-CoA conversion feeding the TCA cycle (Duarte et al, 2021). Additionally, consistent with the lysine degradation pathway, TZD treatment associates with depleted carnitine and its precursor, γ-butyrobetaine. Given carnitine’s central role in transporting long-chain fatty acids into mitochondria for β-oxidation (carnitine shuttle), this reduced carnitine pool provides a novel mechanistic explanation for the observed perturbation in cardiac energetics following TZD administration, as we described in our previous paper (Al Sultan et al, 2024). While both medications elicited significant effects on branched-chain amino acid metabolism, our study additionally highlights pronounced modulations in the aromatic amino acid pool, particularly L-phenylalanine and L-tyrosine. Despite their marginal contribution as energy substrates, these observations echo prior reports linking such alterations to cardiac remodelling (Geng et al, 2020; Karwi & Lopaschuk, 2023). Nevertheless, the ability of these amino acid fingerprint changes to serve as early biomarkers for subclinical cardiac hypertrophy in the context of acute TZD administration remains elusive and necessitates further research.
TZD treatment of AC16 cells triggered a metabolic shift towards anaerobic glycolysis, orchestrated by the upregulation of key glycolytic enzymes and glucose transporters, such as aldolase A and lactate dehydrogenase A noted in TZD-treated cells. Additionally, overexpression of pyruvate dehydrogenase suggested a compensatory mechanism to decrease mitochondrial oxygen consumption and potentially suppress the TCA cycle. These observations collectively indicate TZD-induced cellular hypoxia, which, along with potential cardiac energy deficits, may drive the observed upregulation of purine metabolism in both drug-treated groups (Doigneaux et al, 2020). The altered expression of Inosine-5'-monophosphate dehydrogenase 1, a key enzyme in de novo guanine nucleotide synthesis (Liu et al, 2023), could explain elevated guanine metabolite levels. Furthermore, upregulation of the purine salvage pathway, as indicated by increased inosine and hypoxanthine, suggests a cellular response to mitigate energy deficits by recovering nucleotides from RNA and DNA degradation (Johnson et al, 2019). Finally, modulated expression of adenylate kinase 6, an enzyme involved in maintaining the nuclear adenine nucleotide pool (Deline et al, 2021), further supports the notion of increased cellular demand for nucleotides under hypoxic and glucose-deprived conditions. Notably, the aforementioned observations regarding perturbations in mitochondrial energetics align with our previously reported in vitro cytotoxicity finding of a significant depletion in mitochondrial ATP upon TZD exposure, underscoring the consistency of these observations and highlighting the potential impact of TZDs on cellular energy production (Al Sultan et al, 2024).
TZD-induced alterations in cardiomyocyte fatty acid synthesis, β-oxidation, and amino acid and purine metabolism, as previously described, further translate to modulations in cellular redox status, highlighting the multifaceted impact of TZD on energy metabolism. Our analysis revealed a disrupted glutathione (GSH) system upon TZD administration, evidenced by a significant decrease in GSH content. This depletion could potentially stem from elevated reactive oxygen species (ROS) generated due to TZD-induced mitochondrial damage. Further proteomic investigation of TZD-treated cells unveiled perturbations in GSH anabolism, contributing to the diminished intracellular GSH pool. Specifically, downregulation of key enzymes was observed: (i) glutathione synthetase, responsible for the rate-limiting step of GSH synthesis (Tan et al, 2023), and (ii) glutathione disulfide (GSSG) reduction-related enzymes, such as glutathione reductase and glucose-6-phosphate dehydrogenase, crucial for recycling oxidised glutathione (GSSG) back to GSH (Tan et al, 2023). These findings collectively suggest a TZD-induced imbalance in the cellular redox state, rendering AC16 cells more susceptible to ROS damage. Further elucidating the role of oxidative stress in TZD-mediated cytotoxicity, we utilised the fluorogenic dye H2DCFDA, a broad-spectrum ROS marker, to quantify intracellular ROS levels in AC16 cells upon TZD exposure. Notably, both PGZ and ROSI induced significant ROS elevation at concentration ranges of 10–100 µM and 1–100 µM, respectively, further supporting the implication of oxidative stress in TZD mediated cytotoxicity. Comprehensive descriptions of the experimental procedures and the corresponding figures are provided in the supplemental materials (Section 1.3, Figure S4).
Building upon established biomarkers and leveraging the power of data-driven-based analysis, this study successfully identified key molecular signatures associated with TZD-induced cardiotoxicity across diverse omics datasets using the DIABLO model and the tune.block.splsda function. Prominent among these were signatures of amino acids such as L-ornithine, L-tyrosine, and glutamine, known HF biomarkers, further solidifying their potential utility in clinical settings (Geng et al, 2020; Karwi & Lopaschuk, 2023). Similarly, proteomic signatures revealed alterations in energy metabolism pathways (OXPHOS, pentose phosphate pathway, fatty acid synthesis) reflected by proteins such as Q16795, P52209, and P49327, respectively. Interestingly, ROSI datasets yielded distinct protein signatures enriched in energy metabolism (e.g. ATP synthase) but additionally highlighted disruption of protein synthesis machinery (P0DN37, P0DN26, Q9Y536), suggesting potential endoplasmic reticulum stress and impaired protein export. Notably, these DIABLO-derived signatures align well with the observed metabolic shifts in AC16 cells upon TZD treatment. However, for their translation into clinically relevant prognostic tools for TZD cardiotoxicity and early detection of subclinical hypertrophy, rigorous validation and further investigation are warranted.
4.2 Limitations and Future Research Directions
While the present investigation has yielded valuable insights into our understanding of TZD cardiotoxicity, it is prudent to acknowledge several limitations before drawing definitive conclusions. Motivated by the National Research Council’s (NRC) emphasis on minimising animal experimentation, this study embraced the growing trend of utilising in vitro-to-in vivo extrapolation methodologies in mechanistic toxicology research (Krewski et al, 2020). Accordingly, AC16 cells were chosen as a relevant cell model for investigation. Recognising the AC16 model’s prominent position within the field of cardiac research and its inherent advantages in terms of growth rate and cost-efficiency relative to other models, this study employed this cell line for its investigations. Despite the strengths of the AC16 model, certain limitations warrant consideration. Namely, its dependence on glycolysis, fibroblast-like morphology, and potential for dedifferentiation, along with the complexities of maintaining differentiated cultures (Davidson et al, 2005), restricted our investigation to proliferative cells, as we previously described in (Al Sultan et al, 2024).
The present study utilises a multi-omics framework, integrating toxicoproteomics and toxicometabolomics analyses, to establish novel causal relationships spanning various molecular levels with unprecedented precision. This comprehensive approach offers significant advantages over single-omics analyses in elucidating the complex interplay between molecular alterations and phenotypic manifestations. However, inherent challenges associated with multi-omics studies, such as variations in technology sensitivity across different investigations and the lack of standardised protocols for sample preparation and data acquisition, can hinder the comparability and reproducibility of findings. Addressing these fundamental limitations is crucial to maximising the advancement and fruitful progress within omics research.
In conclusion, this study pioneers the integration of LC–MS-based toxicoproteomics and toxicometabolomics data to unravel the mechanistic underpinnings of TZD-induced cardiotoxicity. The network analysis of proteo-metabolomic data revealed a distinct fingerprint of perturbed biochemical pathways, primarily involving energy metabolism. Additionally, the study identified a marked disruption in the GSH system, indicating an imbalanced redox state triggered by TZD administration. These findings collectively illuminate promising therapeutic targets, paving the way for future research to improve the safety profile of TZD agents.
{kind=link}