Senescence-related feature biomarkers identified using an ML algorithm
Univariate analysis identified 27 genes associated with prognosis among the 296 SRGs obtained from CellAge (Fig. 1A). Subsequently, using two distinct ML algorithms—LASSO regression and SVM-RFE—we searched TCGA dataset for candidate GC diagnostic markers. LASSO regression enabled the reduction of differentially expressed genes (DEGs), revealing nine genes as diagnostic indicators of GC (Fig. 1B). Furthermore, 10 feature genes were discovered in DEGs using the SVM-RFE method (Fig. 1C). Eight diagnostic feature genes were determined via the intersection of the two algorithms (Fig. 1D). Finally, four genes with AUC values exceeding 0.9 were selected for further investigation: PDGFRB, PEX5, HIF1A, and FEN1.
Four SRG regulators in GC and their genetic variability
The role of four SRGs in GC was investigated, considering copy number variations (CNVs). The chromosome sites depicting CNV alterations in the four SRGs are illustrated in Fig. 2A. Examination of PDGFRB, PEX5, HIF1A, and FEN1 indicated a high prevalence of CNV mutations, with widespread amplification observed (Fig. 2B). Further analysis demonstrated overexpression of the four SRGs in GC samples (Fig. 2C). KM analysis (Fig. 2D-G) revealed that low expression of HIF1A and PDGFRB significantly increased OS in TCGA dataset, whereas low expression of PEX5 and FEN1 remarkably reduced OS compared to high expression. Thus, both genetic variation and expression in these four SRGs were identified as critical factors in the course of GC.
Single-cell correlation between four SRGs and the GC TIME
Senescence has an impact on immune cell infiltration into the TIME(18). Utilizing the TISCH database in STAD_GSE167297, we examined the single-cell expression of the four SRGs in the GC TIME. The Violin Chart in Fig. 3A illustrates high expression of the four SRGs, especially HIF1A, in CD8T cells, B cells, DC cells, mast cells, and macrophages/monocytes. Figures 3A-F show the expression of the four SRGs in various immune cell clusters, providing strong evidence of a link between the expression of these SRGs and immune cell infiltration in GC.
Development of a senescence-related signature
The univariate Cox regression model, processed using LASSO Cox regression, provided coefficients for the four SRGs selected based on minimal requirements (Fig. 4A, B). Subsequently, a quantitative indicator, riskScore, was derived using the formula: riskScore = (− 0.2029 × FEN1 expression) + (0.2358 × HIFA expression) + (0.2047 × PDGFRB expression) + (− 0.4524 × PEX5 expression). Using this formula, the riskScore for each patient was calculated, and two distinct groups (high-risk, n = 185; low-risk, n = 186) were obtained based on an optimal threshold value of 0.7752. Principal component analyses (PCA) revealed two distinct components for the two risk groups (Fig. 4C). Examination of clinicopathological characteristics indicated significant differences between high- and low-risk groups in terms of T stage, histologic grade, and pathologic stage in the TCGA cohort (Fig. 4D). KM analyses demonstrated a significantly longer OS in low-risk patients than in high-risk patients, whether using data from TCGA (Fig. 4E) or GEO (Fig. 4F) cohorts.
Verification of the senescence-related signature in the GEO dataset
To confirm the consistency and accuracy of the results, the riskScore model was applied to 431 patients with GC in the GEO dataset. Two subsets of TCGA data were generated using the same threshold value (= 0.7752): low-risk (193 patients) and high-risk (238 patients). Heatmaps of the four SRG expression patterns, riskScore distribution, and survival status of patients in TCGA-GC and GEO datasets demonstrated a similar trend (Figs. 5A-F).
Clinical significance of the senescence-related signature in the GEO and TCGA datasets
To assess the therapeutic implications of senescence, we examined the significance of the clinicopathological factors and riskScore. Univariate Cox regression analysis of TCGA-GC and GEO datasets revealed riskScore and pathological stage as significant risk factors (Fig. 6A, E). The riskScore functioned independently as a reliable prognostic marker in both TCGA-GC (hazard ratio (HR) = 2.335 (1.550–3.519); p = 0.001) (Fig. 6B) and GEO datasets (HR = 1.454 (1.124–1.881); p = 0.004) (Fig. 6F) according to multivariate Cox regression. KM analyses confirmed that the survival of low-risk patients was not significantly different from that of high-risk patients in early disease stages (Fig. 6C, G) but was significantly prolonged in late disease stages compared to high-risk patients (Fig. 6D, H).
Association between senescence-related signatures, somatic mutation, and immunological state
The formation of tumors is often triggered by mutation accumulation(19). The difference between low- and high-risk somatic mutations was investigated (Fig. 7A-B), revealing more pronounced immune-associated alterations in the high-risk population. In the high-risk group, TTN (46%), TP53 (37%), MUC16 (24%), LRP1B (20%), and ARID1A (23%) exhibited the highest mutation frequencies, whereas in the low-risk group, TTN (54%), TP53 (47%), MUC16 (36%), LRP1B (33%), and ARID1A (27%) showed the highest mutation frequencies. Patients were classified into high- and low-TMB groups based on the median TMB value, revealing a negative correlation between life expectancy and TMB levels in patients with GC (Fig. 7C, p = 0.020). Four distinct categories were used to divide the patients as per their RiskScore and the corresponding TMB threshold: high-TMB + low-risk, high-TMB + high-risk, low-TMB + high-risk, and low-TMB + low-risk. Prolonged OS rates were observed in the low-TMB + low-risk group compared to the high-TMB + high-risk group (Fig. 7D, p = 0.005). Since somatic mutations are strongly correlated with the immune microenvironment(20), the relationship between riskScore and immune function was explored. Using ssGSEA, the level of enrichment for various subsets of immune cells was determined, revealing higher infiltration levels of immune cells in high-risk patients, except for CD4 + T cells and neutrophils (Fig. 7E). Additionally, ssGSEA findings from TCGA-GC datasets suggested that the high-risk group exhibited enrichment most immune-associated functions (Fig. 7F). The TIDE score, a tool predicting the likelihood of tumor cells evading the immune system, was remarkably lower in the low-risk group, indicating greater potential benefit from immunotherapy (Fig. 7G).
Further validation of the connection between senescence-related signatures and immune status in single-cell sequencing
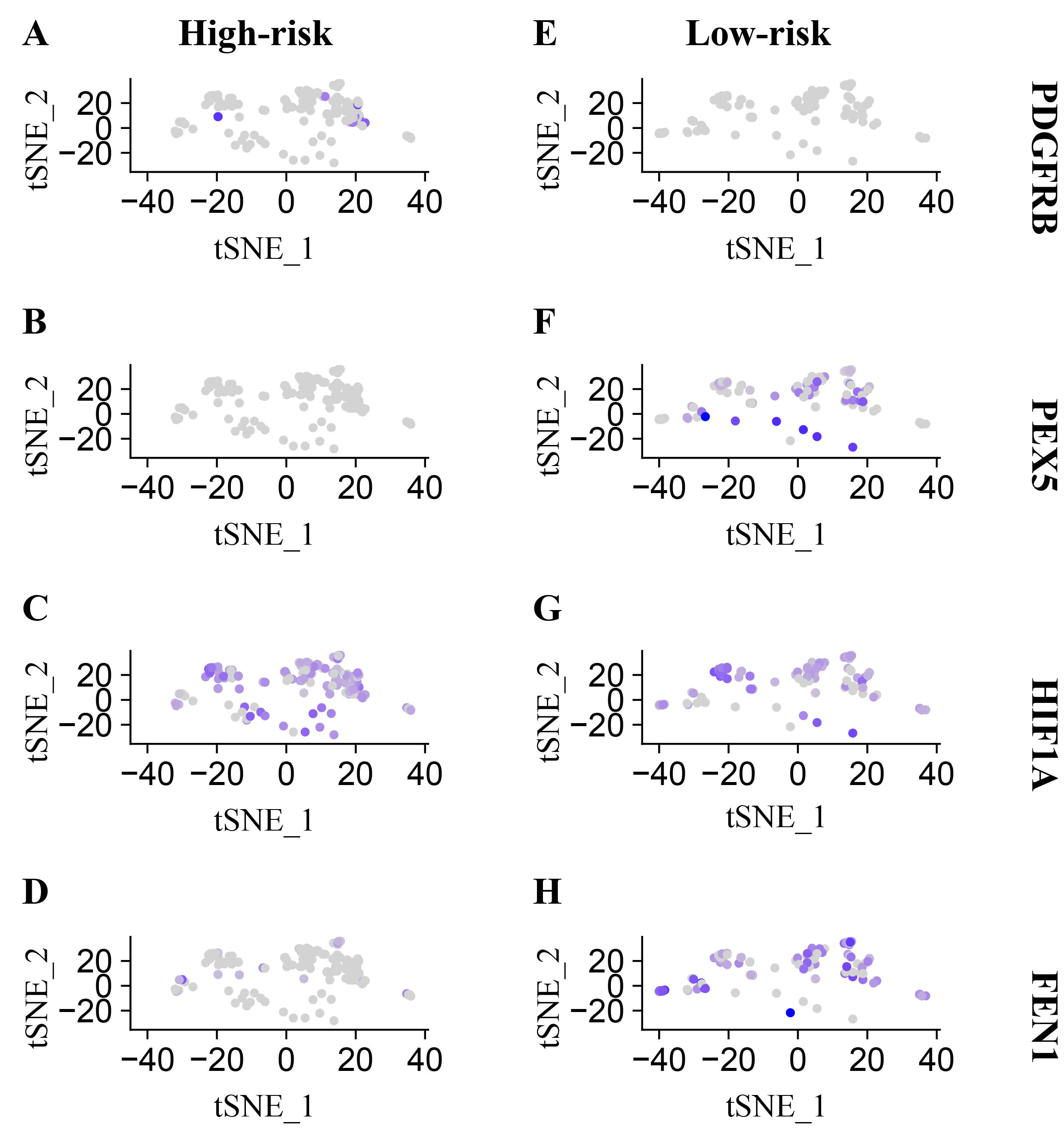
The preceding results were corroborated using single-cell sequencing, wherein 22,270 single cells were isolated after a rigorous quality check. The expression of the four SRGs in single-cell sequencing at high and low risk aligned with previous findings, showcasing upregulation of PDGFRB and HIF1A and downregulation of PEX5 and FEN1 in patients at high risk relative to those at low risk (Figure S2). Employing PCA for dimensionality reduction, the cells were categorized into 10 distinct clusters, including monocytes, neutrophils, CD8 + T cells, macrophages, T helper cells, CD4 + T cells, epithelial cells, mast cells, B cells, cancer cells, and endothelial cells (Fig. 8A, B). Consistent with prior research, the high-risk individuals exhibited increased levels of immune cells including monocytes, neutrophils, and CD8 + T cells, whereas the low-risk individuals showed a greater concentration of epithelial, cancer, and endothelial cells (Fig. 8C). The differences in cell proportions were statistically significant (Fig. 8D-I).
Sensitivity of senescence-related signatures to targeted therapy and chemotherapy
The primary treatment modalities for advanced GC include chemotherapy, immunotherapy, and targeted therapy(21). Given the established value of senescence-related signatures in predicting patient prognosis in advanced GC, we examined the association between riskScore and responses to chemotherapy and targeted treatments. Using the R package “OncoPredict,” we determined the IC50 for various chemotherapeutic and targeted drugs. We found that the IC50 values for sorafenib, bortezomib, gemcitabine, dabrafenib, oxaliplatin, lapatinib, cytarabine, gefitinib, paclitaxel, and 5-fluorouracil were higher in individuals at high risk than in those at low risk, suggesting lower sensitivity to these treatments (Fig. 9A-F). Thus, individuals at low risk might respond better to conventional chemotherapy and targeted therapies.
Significant gene sets between low- and high-risk groups identified via GSEA
GSEA identified several significant gene sets enriched in the high- and low-risk groups from TCGA-GC data. In the high-risk group, the calcium signaling pathway, hematopoietic cell lineage, hypertrophic cardiomyopathy, focal adhesion, and dilated cardiomyopathy were enriched (Fig. 10A). Conversely, the low-risk group exhibited enrichment in gene sets associated with cell cycle(22), DNA replication(23), glycerolipid metabolism(24), ribosomes(25), and spliceosomes(26) (Fig. 10B), all of which are associated with senescence.
Validation of SRG expression and alteration
To validate SRG expression (FEN1, HIF1A, PDGFRB, and PEX5) qRT-PCR was conducted. The findings demonstrated higher relative mRNA expression in GC tissues than in paracancerous tissues for FEN1, HIF1A, PDGFRB, and PEX5 (Fig. 11A-B). Consistent with the results for mRNA expression, the Human Protein Atlas (https://www.proteinatlas.org/) confirmed elevated protein levels of FEN1, HIF1A, and PDGFRB in GC tissues compared to normal tissues, except for PEX5 (Fig. 11C).
{kind=link}
{kind=link}