Because of the contradiction between the increased PA content and the decreased FASN expression in some cells of the lung tissue, we first measured the expression of FASN in lung fibroblasts of IPF and bleomycin-induced fibrotic mice. The double-labeled immunofluorescence staining showed that FASN was co-localized with α-SMA positive fibroblasts in the fibrotic lesions of IPF and bleomycin-induced fibrotic mice. Furthermore, TGFβ1 stimulation significantly increased the expression of FASN in MRC-5 cells. In addition, compared with control mouse fibroblasts, the expression of FASN in fibroblasts isolated from bleomycin-induced fibrotic lungs was upregulated too. This discovery suggested that the increased expression of FASN in lung fibroblasts may contribute to the elevation of PA content in IPF lung tissue.
As we all know, the aberrant activation of β-catenin in fibroblasts is crucial in promoting cell proliferation, migration, and fibroblast to myofibroblasts transformation in IPF [23]. In other words, all changes in the fibroblasts triggered by β-catenin activation require more nutrients and consume more energy. Previous studies suggested that the nutrient and energy requirements of the cells were crucial factors in promoting the expression of FASN [24, 25]. Therefore, we examined whether β-catenin promoted the expression of FASN in both MRC-5 cells and HEK293T cells. The results found that overexpression of β-catenin significantly promoted the protein and mRNA of FASN. This indicated that except for TGF-β1, β-catenin was also a crucial factor in promoting the expression of FASN.
Previous studies have shown that invasive fibroblasts drive the disease progression of IPF [26, 27]. Under normal conditions, lung fibroblasts reside in the interstitial spaces between the alveolar and capillary basal laminae [28]. After lung injury, a population of lung fibroblasts may migrate or invade through alveolar basement membranes [28, 29]. Targeting inhibition of lung fibroblasts invasive has shown the curative effect of treatment for lung fibrosis [30, 31]. For instance, fibroblasts from IPF patients with high programmed cell death ligand 1 (PDL1) expression showed greater migration and invasion abilities. Targeting PDL1 in fibroblasts by CRISPR knockout or anti-PDL1 neutralizing antibodies significantly inhibited fibroblast invasion in vitro and alleviated pulmonary fibrosis in vivo [32]. The pathway analysis of invasive and non-invasive fibroblasts from IPF patients and healthy donors found that the activation profile of aggressive fibroblasts was very similar to that of lung adenocarcinoma cells, and human epidermal growth factor receptor 2 (HER2) was the most obvious, while antagonistic HER2 signaling attenuated fibroblast invasion and ameliorated pulmonary fibrosis [33]. Interestingly, past studies have proved that when FASN was phosphorylated by HER2 on Tyr66, this phosphorylation of FASN increased the activity of the metalloproteinase MMP-9, leading to cell invasion [34]. In the present study, both knockdown FASN with FASN shRNA and pharmacological inhibitor C75 remarkably reduced migration and invasion of fibroblasts. This proved that the inhibition of FASN was very helpful for the therapeutic management of IPF.
In addition, the uncontrolled proliferation of lung fibroblasts accumulated in fibroblastic foci is considered the hallmark of cells in the progression of pulmonary fibrosis [35]. The abnormal proliferation of fibroblast was a significant factor in promoting focal fibrosis expansion [36]. Many profibrotic mediators including interleukin IL-1β, TGF-β, and their cascades, have been identified to stimulate the proliferation of fibroblasts [37, 38]. In the past years, although the proliferation of fibroblasts was not studied as a single target, many studies have proved that the proliferation ability of fibroblasts was significantly inhibited when the migration and the differentiation of fibroblasts were inhibited [39, 40]. This suggests that the function of fibroblast migration and activation may be closely related to its proliferation ability. Coincidentally, studies have demonstrated that FASN promoted cell proliferation, migration, and invasion [41]. For instance, in non-small cell lung cancer, FASN induced epidermal growth factor receptor palmitoylation by inducing PA synthesis, promoting signal transduction, and leading to cell proliferation [42]. In adrenocortical carcinoma, overexpression of miR-486-3p led to the predicted target gene of FASN and its downstream product PA decreasing then reduced the cell clone growth [43]. In the present study, both the knockdown FASN with shRNA and pharmacological inhibitor C75 remarkably reduced the proliferation of fibroblasts. This proved that the inhibition of FASN may be an effective way to inhibit the expansion of fibrotic foci and prevent the progress of pulmonary fibrosis.
The lineage tracing studies of the origin of myofibroblasts in pulmonary fibrosis have revealed that pulmonary fibrosis lacked or rarely experiences epithelial-mesenchymal transition, and resident lung fibroblasts expressing collagen 1 were the main source of myofibroblasts [44]. In this study, FASN inhibition significantly reduced the expression of collagen 1, which will contribute to reducing the source of myofibroblast in pulmonary fibrosis. More importantly, abnormal deposition of extracellular matrix (ECM) proteins is a crucial factor in the progress of lung fibrosis remodel, which results in impaired lung function and clinical symptoms of the respiratory system in IPF [45, 46]. Myofibroblasts produce large amounts of ECM components (such as collagen and fibronectin) [45]. In the present study, the FASN shRNA and C75 markedly reduced the protein and the mRNA expression of fibronectin, collagen 1, and α-SMA. This suggested that FASN inhibition may serve as an effective means to reduce ECM deposition in the lung.
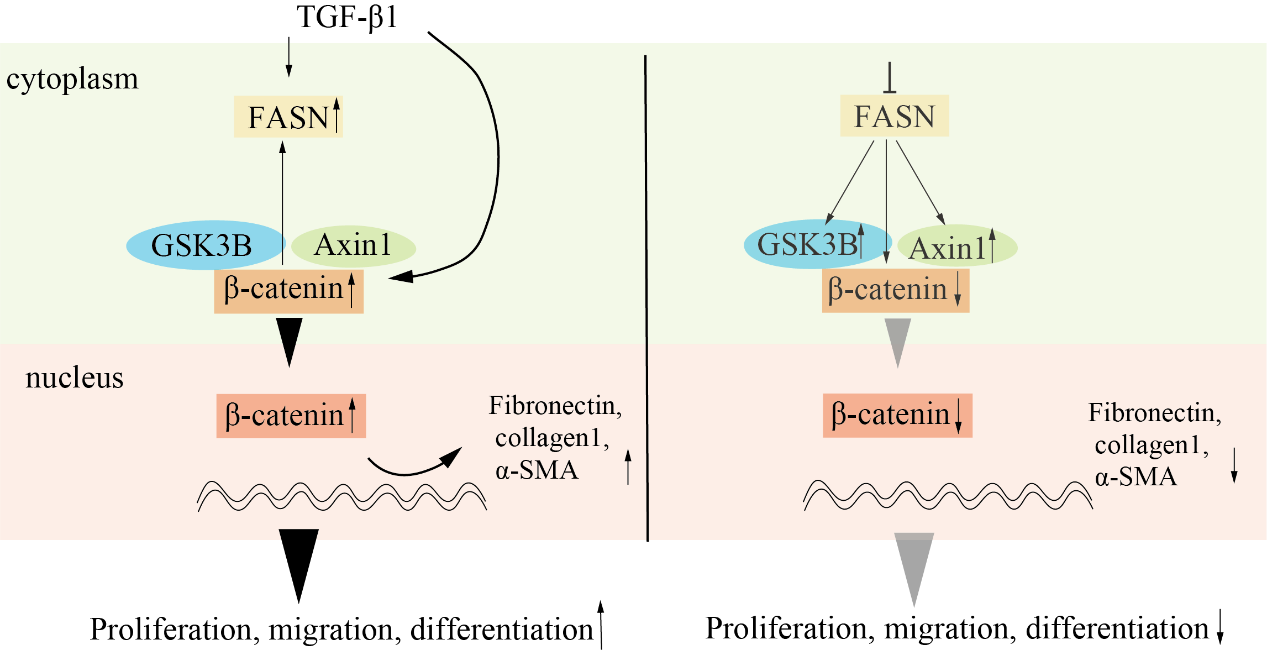
It is well known that the abnormally activated Wnt/β-catenin signaling pathway in fibroblast plays a crucial role in driving lung fibrosis [47, 48]. After various lung injuries in vivo and in vitro, β-catenin as the central mediator accumulated in the cytosol and translocated to the nucleus, where it formed a complex with lymphoid enhancer-binding factor 1 to regulate transcription [49]. As we all know, FASN increases nucleic acid, protein, and lipid synthesis to provide energy to the cancer cell, and also involved in various functions such as immune evasion, resistance to cell death, and so on [41]. Previous studies have also suggested that a strong link existed between FASN and the Wnt/β-catenin signaling pathway. For example, fatty acid binding protein 5 directly interacted with FASN and regulated its expression, thereby promoting lipid droplet deposition and activating the WNT/β-catenin signaling pathway [50]. In salivary adenoid cystic carcinoma cells, FASN inhibition attenuated invasion, metastasis, and epithelial-mesenchymal transition in a PRRX1/Wnt/β-catenin dependent manner [51]. But the exact mechanism was still not known. In the present study, FASN shRNA or C75 obviously reduced the total protein content and mRNA level of β-catenin in fibroblasts. This suggests that the inhibition of FASN regulated the expression of β-catenin at the transcriptional and protein level, respectively. Furthermore, by separating the cytoplasm and nucleus in MRC5 cells, we found that the FASN shRNA or C75 cut down the content of β-catenin both in the cytoplasm and nucleus.
To further clarify the exact mechanism of why FASN inhibited the expression of β-catenin and β-catenin promoted the expression of FASN, we hypothesized that β-catenin or some negative regulators of the Wnt/β-catenin pathway (GSK3B, CK1a, and Axin1) interacted with FASN, the IP results found in HEK293T cells, β-catenin, GSK3B and Axin1 interacted with FASN. Further experiments showed that inhibition of FASN by shRNA and C75 stably increased the expression of GSK3B and Axin1. It was consistent with the result that inhibition of FASN reduced the expression of β-catenin.
Next, to observe the specific effect of FASN inhibition on pulmonary fibrosis, we treated bleomycin-induced fibrotic mice by intraperitoneal injection of C75. The results showed that C75 significantly reduced the protein level of fibronectin, and vimentin, and β-catenin, ameliorated the structural changes in bleomycin-induced pulmonary fibrosis.
However, previous studies have found that specific knockout FASN in AECII worsen pulmonary fibrosis [52]. and specific overexpression FASN in AECII reduced bleomycin‑induced lung injury/fibrosis in bleomycin-induced mice lung fibrosis [53]. These results seem to contradict the present study. But our results give a reasonable explanation by lucky coincidence. Because the expression of FASN in AECII of IPF lung was decreased [53], the decreased expression of FASN may inactivate the Wnt/β-catenin signaling pathway, which led to AECII lost its stemness and failed to repair damaged AECI. On the contrary, the overexpression of FASN in AECII may reactivate β-catenin, and successfully fix the damaged AECI, thereby alleviating pulmonary fibrosis.
Overall, we found the expression of FASN was increased in fibroblasts of fibrotic lungs, the inhibition of FASN reduced the activity of the Wnt/β-catenin signaling pathway, effectively alleviating pulmonary fibrosis. Therefore, the inhibition of FASN in fibroblasts in pulmonary fibrosis is a potential therapeutic approach for pulmonary fibrosis.
CONFLICT
OF INTRERESTS
The authors declare that they have no conflict interests.
{kind=link}