3.1 Clincal samples collection
Labial gland tissues were collected from 15 female SS patients diagnosed at the Oral and Maxillofacial Surgery Department of the Ninth People's Hospital of Shanghai. Additionally, control tissues were collected from 15 HDs. All patients are agreed to participate in this study and provided informed consent. This research received approval from the Ethics Committee of Shanghai Ninth People's Hospital (Approval No.: SH9H-2019-T 159-2).
3.2 Cell culture
The human submandibular tumor cell line A253 (American Type Culture Collection, ATCC, Maryland, USA) was cultured in complete high glucose DMEM/F-12 medium (11320033, Gibco, New York, USA). The culture medium was supplemented with 10% fetal bovine serum (FBS, Gibco). Cells were maintained in a humidified incubator at 37°C with 5% CO2. Experiments were conducted when cells reached 60-70% confluence. Various organic compounds used for cell treatment were added to this culture medium. The drugs used were as follows: diABZI STING agonist (Compound 3) (S8796, Selleck), 100nM, incubated for 6 hours; H-151, 1μM, incubated for 6 hours; 4-PBA (S3592, Selleck), 3mM, incubated for 6 hours; Thapsigargin (S7895, Selleck), 40nM, incubated for 3 hours (19). For siRNA transfection, cells were cultured to 40% confluence, then washed and transfected according to the manufacturer's instructions using siRNA and transfection reagent (Lipofectamine 3000, L3000001, Thermo Fisher). Appropriate concentrations of siRNA and P3000, or Lipofectamine 3000 were separately dissolved in serum-free DMEM, then mixed and incubated for 25 minutes before being added to the cells in serum-free DMEM. After 6 hours, the medium was replaced with serum containing DMEM. Cells were collected for necessary assays 24 hours post-transfection.
3.3 Animal experiments
The female NOD/Ltj mice were purchased from the Model Animal Research Center of Nanjing University (China). We initiated intraperitoneal injections at 12 weeks of age, administering a uniform volume of 200μL of either drug solution or 200μL of DMSO. Injections were administered once daily for a duration of three weeks. For mouse treatment with H-151 (S6652, Selleck), referencing the literature, injections were performed at concentrations of 300nM and 750nM (20-22); when treating mice with 4-PBA (S3592, Selleck), the concentration used was 150mg/kg (23); When treating mice with Thapsigargin (S7895, Selleck), the concentration used was 0.2 ng/L (24); when treating mice with Tunicamycin (S7894, Selleck), the concentration used was 150µg/kg(24). After completing the treatment, the mice were euthanized, and SG tissues and serum were extracted. The tissues were fixed, embedded in paraffin, and subjected to Hematoxylin and Eosin (H&E) staining. The severity of glandular tissue lesions was assessed using a widely adopted scoring system (25). A count was conducted on the total lymphocytic foci containing fewer than 50 lymphocytes in each slice. Each group consisted of a minimum of 3 mice, and treatments were administered as directed. Throughout the experiment and daily care, animals received humane treatment following the standards outlined in the "Guide for the Care and Use of Laboratory Animals" published by the National Institutes of Health (NIH) (26). This study received approval from the Ethics Committee of Shanghai Jiao Tong University School of Medicine.
3.4 RNA extraction and real‐time polymerase chain reaction (RT–qPCR)
We extracted total cellular RNA using TRIzol reagent (15596018, Takara Bio, Japan). Following the manufacturer's instructions, we performed reverse transcription of RNA using the PrimeScript RT Kit (Perfect Real‐Time, RR037A, Takara Bio). Subsequently, cDNA obtained from 2 μg of RNA was utilized for real-time PCR on the LightCycler 480 II (Roche, Switzerland) using TB Green Premix Ex Taq (RR420A, TaKaRa Bio). RNA levels were analyzed using LightCycler 480 SW 1.5.1 (Roche). All reactions were conducted in triplicate in a 384-well PCR microplate, and β-actin was used as an internal reference to standardize gene expression levels. Primers were procured from Sangon Biotech. Initially, primer efficiency was assessed using a relative standard curve, and the fold change in RNA expression was calculated as -ΔΔCt. Subsequently, quantitative results were obtained. The primer sequences are listed in Supplemental Table 1.
3.5 Western blotting
After treating the cells with the required drugs, cells were harvested. Total protein was collected using IP lysis buffer (UH290847, Thermo Fisher). After adding sample buffer (NP0007, Thermo Fisher), the mixture was boiled for 10 minutes. The antibodies used are listed in Supplemental Table 2. All antibodies were diluted at 1:1000 according to the manufacturer's instructions. Subsequently, samples underwent a standard protein immunoblot procedure using the Bio-Rad system and gel kit (PE008, Zhonghui Jingcai).
3.6 Apoptosis analysis by Annexin V/PI assays
The SG of mice treated with the injected drugs and subsequently euthanized were gently extracted, taking care to handle them delicately. Next, the submandibular gland tissues were digested in Type II collagenase (1mg:1ml PBS, 17101015, Gibco) for 2 minutes. The digested tissue was then passed through a cell strainer, centrifuged, and the supernatant was discarded. The pellet was resuspended in clean PBS. For the assessment of apoptosis, the resuspended cells were collected in flow cytometry tubes and stained using the membrane protein V/PI assay kit (PL748, Dojindo) according to the manufacturer's instructions. After staining with the epithelial marker antibody CD326 and washing, the cells were stained with the apoptosis detection reagent for 15 minutes. Apoptotic cells were analyzed through flow cytometry (27).
3.7 JC-1-based mitochondrial dysfunction analysis
The JC-1 MitoMP Detection Kits (MT09, Dojindo) were employed to assess mitochondrial function in A253 cells. In brief, 2 μM JC-1 working solution was added 45 minutes prior to collection. Following JC-1 treatment, cells were washed twice with PBS and then imaged. The fluorescence intensity was evaluated under a microscope.
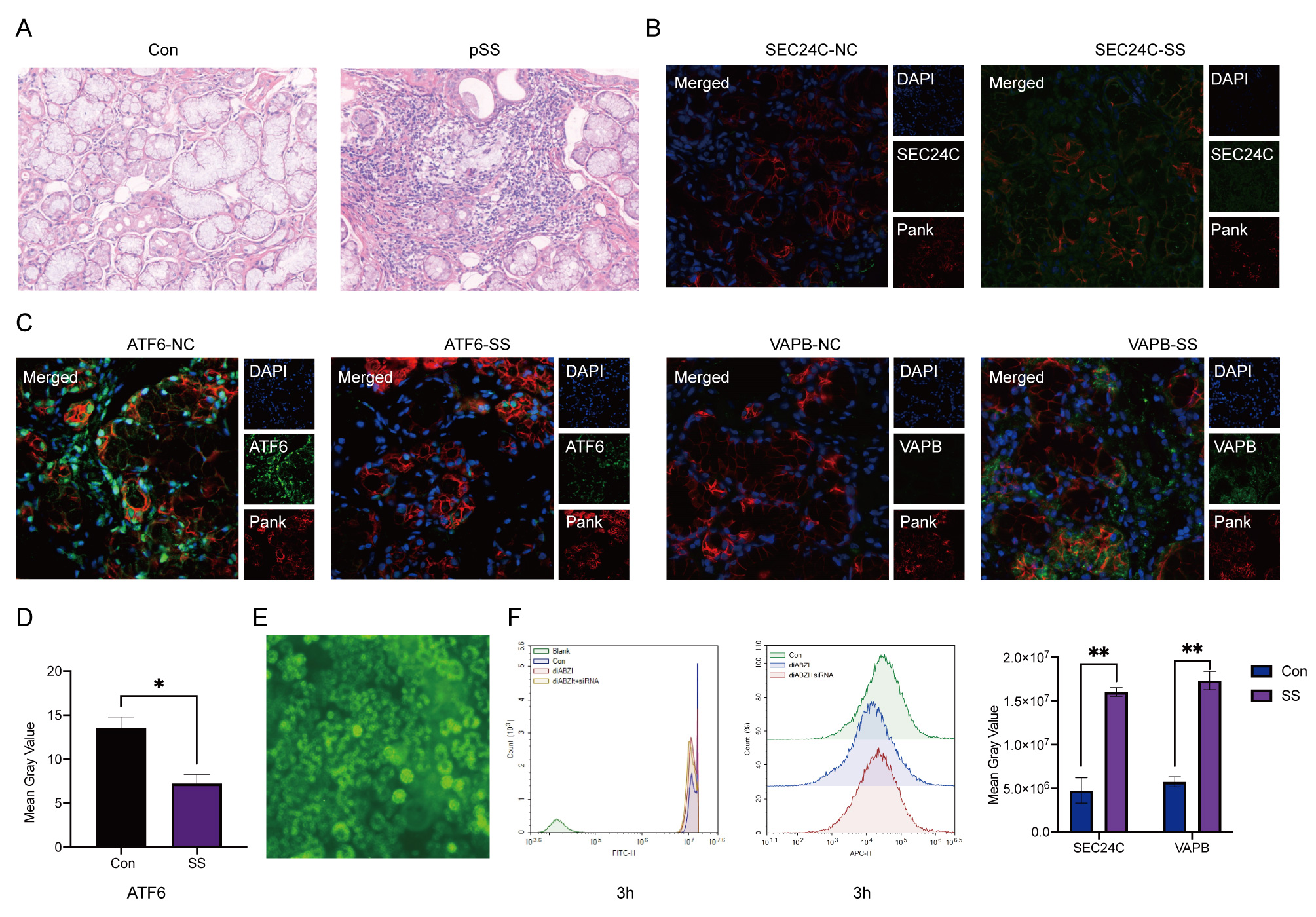
3.8 H&E staining, immunohistochemistry and immunofluorescence
The 4 μm paraffin-embedded sections were stained with H&E or used for immunohistochemistry. The antibodies used are listed in Table 2. Samples were incubated at room temperature with 3% H2O2 for 5-10 minutes to eliminate endogenous peroxidase activity. Cells were then blocked with 5-10% normal goat serum and incubated at room temperature for 10 minutes. Biotin-labeled secondary antibodies, appropriately diluted, were added dropwise and incubated at 37°C for 10-30 minutes. Next, peroxidase-labeled streptavidin, appropriately diluted, was added dropwise and incubated at 37°C for 10-30 minutes. If immunofluorescence is required, proceed with the incubation of fluorescent secondary antibodies.
3.9 ROS assay
A253 cells were seeded in six-well plates and cultured overnight until they adhered. Then, drug treatment was administered. The cell culture medium was replaced with serum-free medium containing 1:1000 DCFH-DA (S0033S, Beyotime), and cells were cultured at 37°C for 20 minutes. Afterward, the cells were washed three times with serum-free medium, digested with trypsin, and prepared into a cell suspension for flow cytometry analysis.
3.10 ES staining, Golgi apparatus staining, Ca2+ staining, fluorescence confocal photography
The IRE1 antibody was used for immunofluorescence staining to label the location of the ER (28). The Golgi staining kit (C1043, Beyotime) and Ca2+ Staining Kit (S1061M, Beyotime) were used according to the manufacturer's instructions. In summary, cells were seeded in confocal dishes and treated with the required drugs, reaching a confluence of about 60%. They were then incubated with serum-free medium containing the staining reagent or the buffer provided by the kit for a specific period of time, typically ranging from 20 minutes to 1 hour, depending on the specific staining kit. Subsequently, cells were fixed with paraformaldehyde for observation and imaging. Regarding changes in Ca2+ concentration in the cytoplasm, we utilized the Ca2+ Concentration Assay Kit (S1061S, Beyotime). After treating the cells, digestion was performed for staining, and flow cytometry was used to detect alterations.
3.11 Immunoprecipitation
After treatment, total protein was extracted from A253 cells. 10 μL of each sample were taken as input, and the rest were incubated with IP magnetic beads (UG288056A, Thermo Fisher Scientific) bound to rabbit IgG antibody overnight at 4。C. On the following day, the beads were discarded, new beads were taken and combined with specific primary antibodies to bind with the samples, followed by another overnight incubation at 4 degrees Celsius. Finally, the beads were retrieved, resuspended in IP lysis buffer and sample loading buffer, boiled for 10 minutes, and the beads were discarded. The samples were then subjected to a standard protein immunoblotting procedure using a Bio-Rad system.
3.12 Transmission electron microscope
First, process the samples by directly digesting the cells and immersing them in electron microscopy fixative. For animal tissues, slice them thinly and fix them in electron microscopy fixative (G1102, Servicebio). Then, observe them using an electron microscope (HT7800, Hitachi).
3.13 Salivary flow rate measurement
Each group of mice received an intraperitoneal injection of 2.4% pentobarbital sodium (5 ml/kg, P-010, Sigma-Aldrich). After anesthesia, mice were injected with pilocarpine (0.125 mg/kg, Sigma-Aldrich). Five minutes later, saliva was collected using dry cotton balls for 10 minutes. The weight of the collected saliva was measured and then subtracted from the weight of the cotton ball. The collected saliva was normalized to the body weight of each mouse.
3.14 Metabolic flux assay
The OCR in A253 cells was analyzed using the XF96 Extracellular Flux Analyzer (Seahorse Bioscience, Massachusetts, USA). Prior to the experiment, 2 × 105 cells were seeded per well on Seahorse 96-well culture plates pre-coated with Cell-Tak (DLW354240, Corning). Under basal conditions, extracellular acidification rate (ECAR) and OCR were measured in XF DMEM containing glutamine, glucose, HEPES, and pyruvate. Reactions to specified concentrations of oligomycin, carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), rotenone, and antimycin A were assessed.
3.15 Statistical analysis
The data was analyzed using GraphPad Prism 9. Paired sample t-tests were performed to compare between-group parameters. All statistical tests were two-tailed, and the significance level was set at p < 0.05.
{kind=link}